In ematologia, il meeting annuale dell’Associazione Europea di Ematologia è uno degli appuntamenti del calendario annuale. L’associazione promuove l’eccellenza nell’assistenza ai pazienti, nella ricerca e nell’educazione in ematologia. Di conseguenza, il congresso trasmette le scoperte più recenti e innovative e i risultati della ricerca sulle malattie ematologiche. Verranno trattati la ricerca clinica e la pratica, la ricerca di base e traslazionale e gli approcci più recenti alla diagnosi e al trattamento.
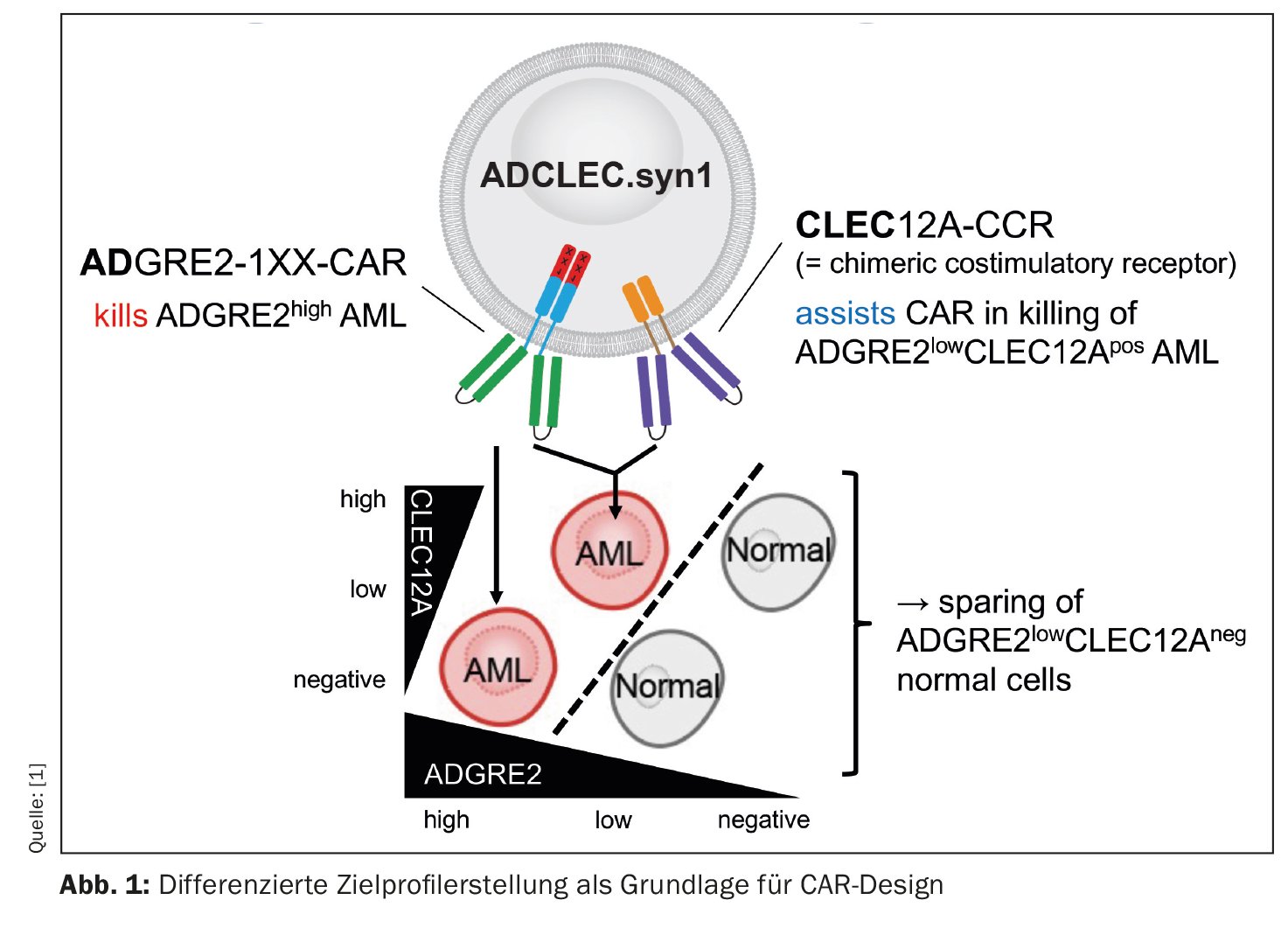
Un gruppo di ricercatori sta studiando una nuova terapia CAR per la leucemia mieloide acuta (AML), i cui risultati preclinici sono incoraggianti [1]. Il concetto chiamato ADCLEC.syn1 utilizza recettori cooperativi che hanno come target ADGRE2 e CLEC12A. In questo modo, si dovrebbe eliminare l’AML e ridurre al minimo le tossicità ematologiche. Questo è stato dimostrato da una serie completa di modelli di efficacia e tossicità in vivo. Le terapie CAR per l’AML incontrano ostacoli a causa dell’eterogeneità clonale e della somiglianza con la normale emopoiesi precoce, che può portare alla fuga dell’antigene e a tossicità ematologiche. Lo studio attuale ha analizzato l’espressione quantitativa dei bersagli di superficie nella LAM e nei tessuti normali, per determinare le finestre terapeutiche che possono essere sfruttate da nuovi disegni combinatori di CAR. Quindi, è stata sviluppata ADCLEC.syn1, una nuova terapia CAR combinatoria che co-targettizza ADGRE2 e CLEC12A per eliminare in modo selettivo le cellule AML con bassi livelli di ADGRE2 e risparmiare le cellule staminali e progenitrici ematopoietiche normali. (Fig. 1). I ricercatori hanno correlato l’espressione dell’antigene bersaglio con l’efficacia delle cellule T ADCLEC.syn1 e CD33-CAR utilizzando xenotrapianti di AML. I risultati hanno dimostrato che ADCLEC.syn1 ha indotto una remissione duratura in diverse linee cellulari AML umane rappresentative dei fenotipi dei pazienti AML recidivati/refrattari. Tuttavia, i topi a cui erano stati somministrati innesti di AML e ricostituiti con cellule ematopoietiche umane normali hanno risposto solo ad ADCLEC.syn1 ma non a CD33-CAR. Questi risultati evidenziano l’importanza della profilazione quantitativa del target CAR nella LAM. ADCLEC.syn1 è ora oggetto di uno studio clinico di Fase I, primo nell’uomo, per la LAM recidivata/refrattaria.
Progressi all’APL
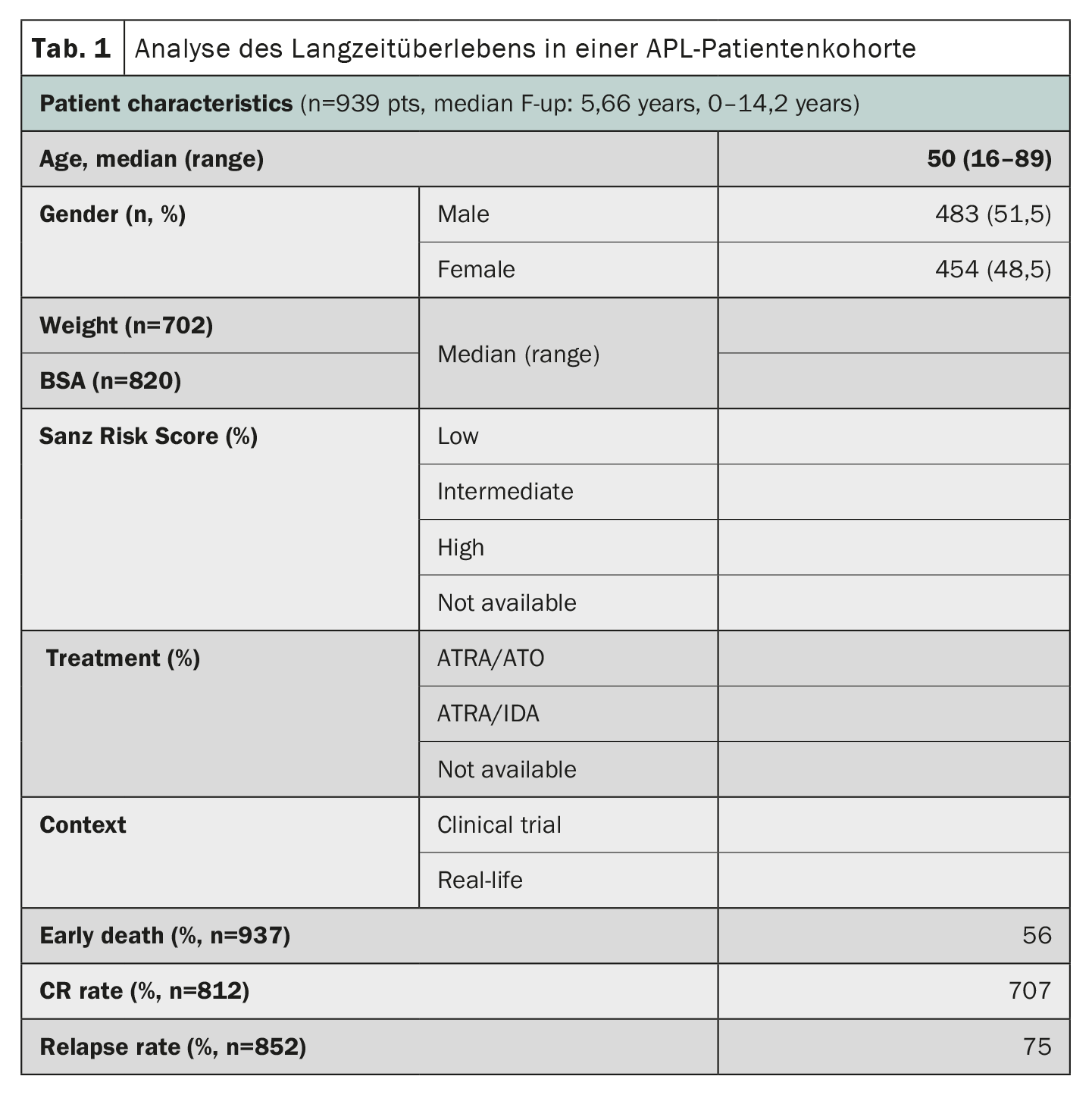
La leucemia promielocitica acuta (APL), un tempo considerata una delle forme più rapidamente fatali di leucemia mieloide acuta, ha mostrato notevoli progressi nei risultati del trattamento. Uno studio che ha utilizzato i dati del registro HARMONY con un’ampia coorte di pazienti ha confermato che la terapia combinata di acido retinoico all-trans (ATRA) e triossido di arsenico (ATO) ha portato a tassi di sopravvivenza globale a 10 anni dell’85-92% nei pazienti con APL [2]. Il registro HARMONY comprende 1868 pazienti con APL, provenienti da due studi clinici e da registri nazionali in sei Paesi, diagnosticati tra il 2007 e il 2020. Di questi, 937 pazienti hanno soddisfatto i requisiti di qualità dei dati e sono stati inclusi nella presente analisi. I dati sono stati armonizzati utilizzando un modello di dati comuni della Observational Medical Outcomes Partnership e registrati nella HARMONY Big Data Platform. I risultati dell’analisi hanno mostrato che i pazienti trattati con il regime ATRA-ATO avevano un tasso di sopravvivenza globale (OS) a 10 anni del 92% rispetto al 75% dei pazienti trattati con il regime ATRA-idarubicina (AIDA) (Tab. 1). Il beneficio in termini di sopravvivenza è stato lo stesso nei diversi gruppi di rischio definiti dal punteggio di rischio Sanz. Anche l’età ha giocato un ruolo importante nella sopravvivenza, con i pazienti più giovani (<50 anni) che hanno ottenuto risultati migliori. Tuttavia, il tasso di decessi precoci (<30 giorni dopo la diagnosi) era simile in entrambi i gruppi (3,4%-5,7%). Nel complesso, questi risultati in un’ampia coorte internazionale di pazienti confermano il significativo beneficio di sopravvivenza della terapia ATRA-ATO senza chemioterapia per i pazienti con APL, indipendentemente dal loro profilo di rischio, e forniscono preziose indicazioni per il trattamento dell’APL.
Guarigione funzionale nella TDT?
I primi risultati intermedi sono incoraggianti [3]: Uno studio di fase III sull’autotempio exagamglogenico (exa-cel), una terapia cellulare non virale, fa sperare in una cura funzionale una tantum per i pazienti con β-talassemia trasfusione-dipendente (TDT). Sono stati evidenziati risultati significativi nell’indipendenza dalle trasfusioni, nel miglioramento dei livelli di emoglobina e nella qualità della vita. Exa-cel riattiva la sintesi dell’emoglobina fetale (HbF) mediante l’editing genico ex vivo CRISPR/Cas9, mirando al gene BCL11A nelle cellule staminali e progenitrici ematopoietiche CD34+ autologhe. Dei 48 pazienti TDT che hanno ricevuto exa-cel, 27 erano valutabili per gli endpoint dello studio all’analisi ad interim prestabilita. I soggetti che sono rimasti indipendenti dalle trasfusioni per ≥12 mesi avevano un tempo medio all’ultima trasfusione di 37 giorni dopo l’infusione di exa-cel e sono rimasti liberi da trasfusioni per 12,1-40,7 mesi. I livelli di emoglobina e gli alleli BCL11A modificati nelle cellule CD34+ del midollo osseo e nelle cellule ematiche nucleate periferiche sono rimasti stabili nel tempo.
Lo studio ha anche osservato miglioramenti significativi nella qualità della vita e un’incisione di neutrofili e piastrine di successo in tutti i pazienti, evidenziando l’efficacia della terapia. Il profilo di sicurezza di exa-cel è stato coerente con il regime di condizionamento mieloablativo a base di busulfano e con le procedure di trapianto autologo, con eventi avversi gestibili. Tutti gli eventi avversi gravi si sono risolti e non ci sono stati decessi, ritiri dallo studio o tumori.
Eritrocitosi incontrollata nella policitemia vera
Lo studio REVIVE ha analizzato l’efficacia del rusfertide, un nuovo mimetico dell’epcidina, nei pazienti con policitemia vera (PV) [4]. Il rusfertide inibisce la produzione di globuli rossi nei pazienti con PV, limitando la disponibilità di ferro. Lo studio ha utilizzato una proteina sintetica simile all’epcidina, che viene normalmente prodotta dal fegato e regola il trasporto del ferro, per trattare l’eritrocitosi associata alla PV (eccessiva produzione di globuli rossi). La fase di astinenza randomizzata di 12 settimane dello studio ha raggiunto l’endpoint primario e ha dimostrato l’elevata efficacia di rusfertide nel controllare l’eritrocitosi, una caratteristica chiave della PV che aumenta il rischio di complicanze tromboemboliche e cardiovascolari.
Lo studio di fase II ha arruolato pazienti con diagnosi di PV secondo i criteri OMS del 2016 e che hanno richiesto un numero eccessivo di flebotomie terapeutiche (TP) in pazienti trattati con TP da solo o con agenti di citoriduzione (CYTO). Rusfertide, somministrato per via sottocutanea una volta alla settimana, è stato aggiunto alla precedente terapia FV. Durante la fase di sospensione, i pazienti sono stati randomizzati a continuare la terapia con rusfertide per 12 settimane o a ricevere un placebo. I dati della fase di ritiro randomizzata hanno dimostrato l’efficacia superiore di rusfertide rispetto al placebo. I pazienti hanno mostrato un tasso di risposta statisticamente significativo (TP-free) del 69,2% rispetto al 18,5% del gruppo placebo. Inoltre, la terapia con rusfertide è stata associata a un controllo sostenuto dell’ematocrito (HCT) rispetto al placebo. Il tasso di assenza di TP nei pazienti ha raggiunto il 92,3%. Il trattamento è stato generalmente ben tollerato, con la maggior parte degli eventi avversi rappresentati da reazioni al sito di iniezione di gravità lieve o moderata, che sono diminuite con il proseguimento del trattamento.
I risultati positivi dimostrano l’efficacia e la tollerabilità di rusfertide come terapia altamente efficace per l’eritrocitosi incontrollata e i sintomi associati nella PV e rappresentano un progresso significativo nel trattamento di questa neoplasia mieloproliferativa maligna. Il composto offre un approccio innovativo basato su un ormone mimetico che colpisce selettivamente l’eritrocitosi incontrollata, fornendo un controllo sostenuto e duraturo dell’HCT e un miglioramento dei sintomi correlati alla PV.
Trattamento dell’anemia nella LR-MDS
Lo studio COMMANDS di fase III ha mostrato risultati promettenti nel trattamento dell’anemia associata alle sindromi mielodisplastiche a basso rischio (LR-MDS) [5]. In un’analisi ad interim pre-pianificata, luspatercept ha mostrato benefici clinici significativi nei pazienti LR-MDS naïve agli ESA rispetto al trattamento standard con epoetina alfa. Questi risultati hanno il potenziale di cambiare il panorama terapeutico dei pazienti LR-MDS che si affidano alle trasfusioni. I pazienti LR-MDS affetti da anemia cronica soffrono di una maggiore morbilità, di un sovraccarico di ferro e di una ridotta sopravvivenza. L’attuale standard di cura, gli agenti stimolanti l’eritropoiesi (ESA), ottengono solo risultati subottimali in questi pazienti. L’analisi ad interim ha valutato l’efficacia e la sicurezza di luspatercept e di epoetina alfa in 356 pazienti LR-MDS ESA-naive, trasfusione-dipendenti. L’endpoint primario dello studio era il raggiungimento dell’indipendenza dalle trasfusioni di globuli rossi (RBC-TI) per ≥12 settimane con un contemporaneo aumento medio dell’emoglobina di almeno 1,5 g/dL durante le prime 24 settimane. Il 59% dei pazienti trattati con luspatercept ha ottenuto un RBC-TI e un aumento concomitante dell’emoglobina, rispetto al 31% del gruppo epoetina-alfa. Il beneficio clinico è stato osservato in tutti i sottogruppi. Anche il raggiungimento degli endpoint secondari era a favore di luspatercept, compreso il miglioramento ematologico. I pazienti con mutazioni genetiche specifiche associate alla MDS, come SF3B1, SF3B1a, ASXL1 e TET2, hanno risposto meglio della media a luspatercept, indipendentemente dal carico complessivo di mutazioni. Luspatercept ha avuto un profilo di sicurezza favorevole, con eventi avversi correlati al trattamento da lievi a moderati. Il tasso di mortalità complessivo era simile nei gruppi luspatercept ed epoetina alfa.
Trattare la LLC in modo efficace
Lo studio CLL14, un’indagine sul trattamento della leucemia linfatica cronica (LLC), ha fornito risultati recenti sugli esiti a lungo termine dei pazienti trattati con venetoclax-obinutuzumab (Ven-Obi) [6]. I risultati dimostrano un’efficacia e una sicurezza sostenute e potrebbero far diventare Ven-Obi l’opzione terapeutica preferita per i pazienti affetti da CLL, compresi quelli con malattia concomitante. In questo studio in corso, 432 pazienti con LLC precedentemente non trattati sono stati assegnati in modo casuale al trattamento con Ven-Obi o con clorambucil-obinutuzumab (Clb-Obi). Dopo un follow-up mediano di 76,4 mesi, Ven-Obi ha mostrato una migliore sopravvivenza libera da progressione (PFS) rispetto a Clb-Obi (PFS mediana 76,2 vs. 36,4 mesi). È importante notare che anche dopo sei anni, il tasso di PFS stimato per Ven-Obi era del 53,1% rispetto al 21,7% per Clb-Obi. Lo studio ha anche dimostrato che Ven-Obi ha comportato un tempo significativamente più lungo al trattamento successivo (TTNT) rispetto a Clb-Obi (TTNT a 6 anni 65,2% vs. 37,1%) (Fig. 2). Questi risultati positivi sono stati osservati in tutti i gruppi di rischio, compresi i pazienti con caratteristiche di CLL ad alto rischio. Inoltre, Ven-Obi ha mostrato un’eccellente risposta alla malattia minima residua (MRD), con il 7,9% dei pazienti senza livelli di MRD rilevabili cinque anni dopo il trattamento, rispetto all’1,9% con Clb-Obi. Non sono stati rilevati nuovi segnali di sicurezza.
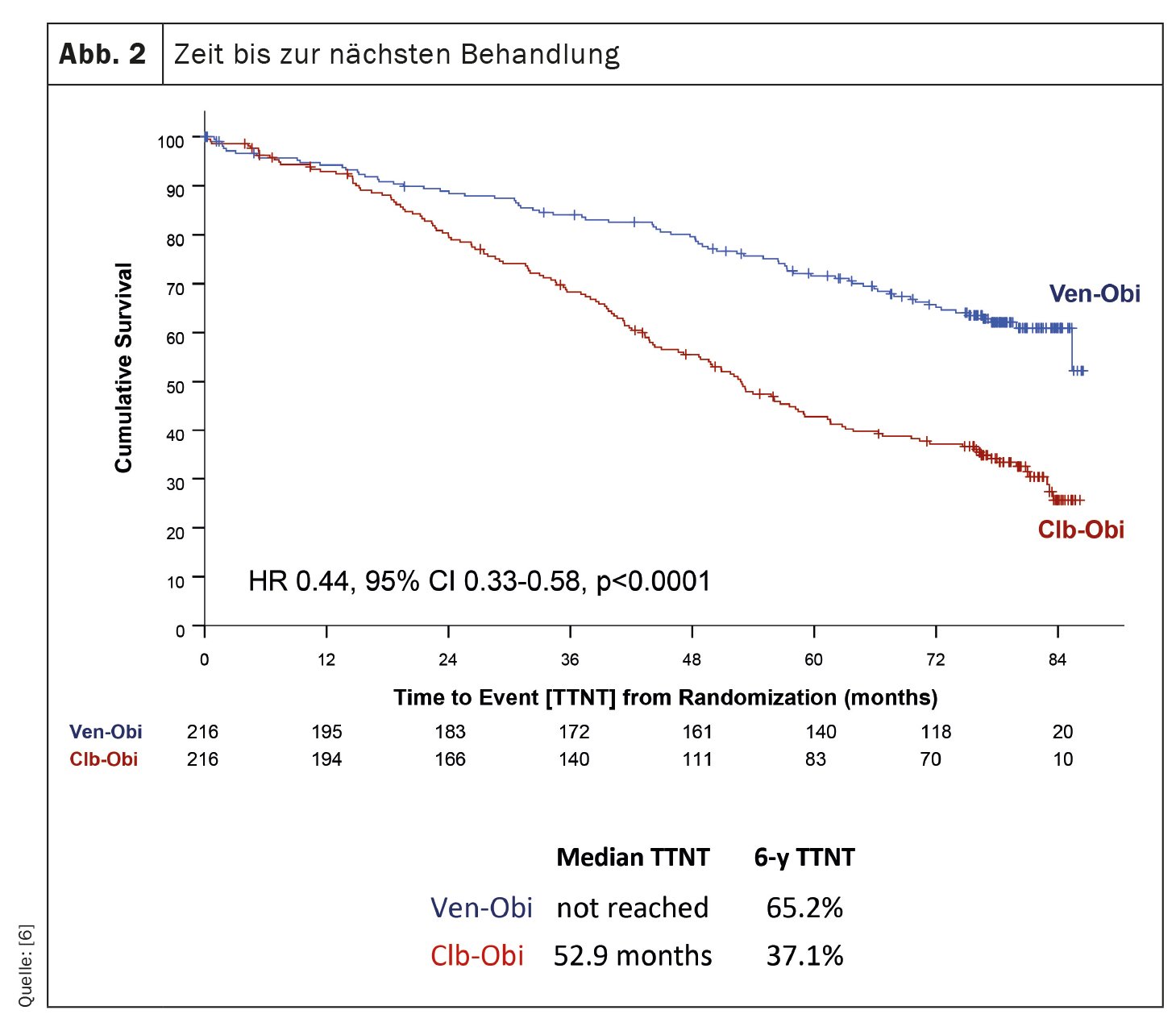
Ven-Obi offre una remissione sostenuta, alti tassi di malattia residua minima non rilevabile e un tempo più lungo per il trattamento successivo. Più della metà dei pazienti è ancora in remissione cinque anni dopo aver completato il trattamento e la maggior parte non ha bisogno di un trattamento di seconda linea.
Emocromatosi ereditaria
L’omozigosi per la variante HFE C282Y causa l’emocromatosi ereditaria, che può potenzialmente portare a diabete, malattie epatiche e cardiache. Uno studio ha ora verificato se l’omozigosi C282Y aumenta il rischio di diabete, malattie epatiche e cardiache, anche nelle persone con ferro plasmatico, saturazione della transferrina o ferritina normali [7]. Inoltre, ha studiato se gli omozigoti C282Y con diabete, malattie epatiche o cardiache hanno un rischio maggiore di morte rispetto ai non portatori di queste malattie. Un totale di 132.542 individui consecutivi di una coorte di popolazione generale sono stati genotipizzati per la variante HFE C282Y e sono stati trovati 422 omozigoti. Gli individui sono stati seguiti in modo prospettico fino a 27 anni. Le informazioni sui contatti ospedalieri provenivano dal registro nazionale dei pazienti, che registra tutti gli ospedali danesi.
Durante il periodo di follow-up, 17 688 persone sono morte per qualsiasi causa, mentre 7702, 2804 e 21 769 persone sono state ricoverate in ospedale rispettivamente per diabete, malattie epatiche e malattie cardiache. Confrontando gli omozigoti C282Y con i non portatori, i rapporti di rischio erano 1,66 per il diabete, 2,16 per le malattie epatiche e 0,98 per le malattie cardiache. Il rischio di diabete era aumentato anche negli omozigoti C282Y con ferro, saturazione della transferrina o ferritina normali (4,35; 1,81-10,48). Gli omozigoti C282Y con diabete avevano un hazard ratio per la morte da qualsiasi causa di 1,94 rispetto ai non portatori con diabete. La percentuale di tutti i decessi negli omozigoti C282Y che potrebbe teoricamente essere evitata se si eliminasse una singola malattia è stata del 27,3% per il diabete e del 14,4% per le malattie epatiche. I risultati suggeriscono che l’attuale strategia di trattamento dell’emocromatosi, che si concentra sull’abbassamento della ferritina, è insufficiente per ridurre il rischio di diabete e il rischio di morte per diabete.
Congresso dell’Associazione Europea di Ematologia (EHA) 2023
Letteratura:
- Hauber S, et al.: Novel CAR Therapy for Acute Myeloid Leukemia Targeting ADGRE2 and CLEC12A Shows Favorable Pre-Clinical Outcomes. S104. 10.06.2023. EHA 2023.
- Voso MT, et al.: ATRA-ATO Regimen Provides Significant Survival Advantage for Patients with Acute Promyelocytic Leukemia. S136. 11.06.2023. EHA 2023.
- Locatelli F, et al.: Exagamglogene Autotemcel: A Potential One-Time Functional Cure for Patients with Transfusion-Dependent β-Thalassemia. S270. 11.06.2023. EHA 2023.
- Hoffman R, et al.: Rusfertide Therapy Serves as a Novel Effective Treatment for Uncontrolled Erythrocytosis in Polycythemia Vera. LB2710. 11.06.2023. EHA 2023
- Della Porta MG, et al.: Luspatercept Is Superior to Epoetin Alfa in Treating Anemia in Lower-Risk Myelodysplastic Syndromes. S102. 10.06.2023. EHA 2023.
- Al-Sawaf O, et al.: Long-Term CLL14 Study Confirms Venetoclax-Obinutuzumab as Effective Treatment for Chronic Lymphocytic Leukemia. S145. 09.06.2023. EHA 2023.
- Mottelson M, et al.: Individuals with HFE C282Y Homozygosity and Diabetes habe almost two-fold risk of death compared to non-carriers with diabetes: A prospective study of a 132,542-individual general population cohort. S280. EHA 2023.
InFo ONKOLOGIE & HÄMATOLOGIE 2023; 11(3): 26–28