
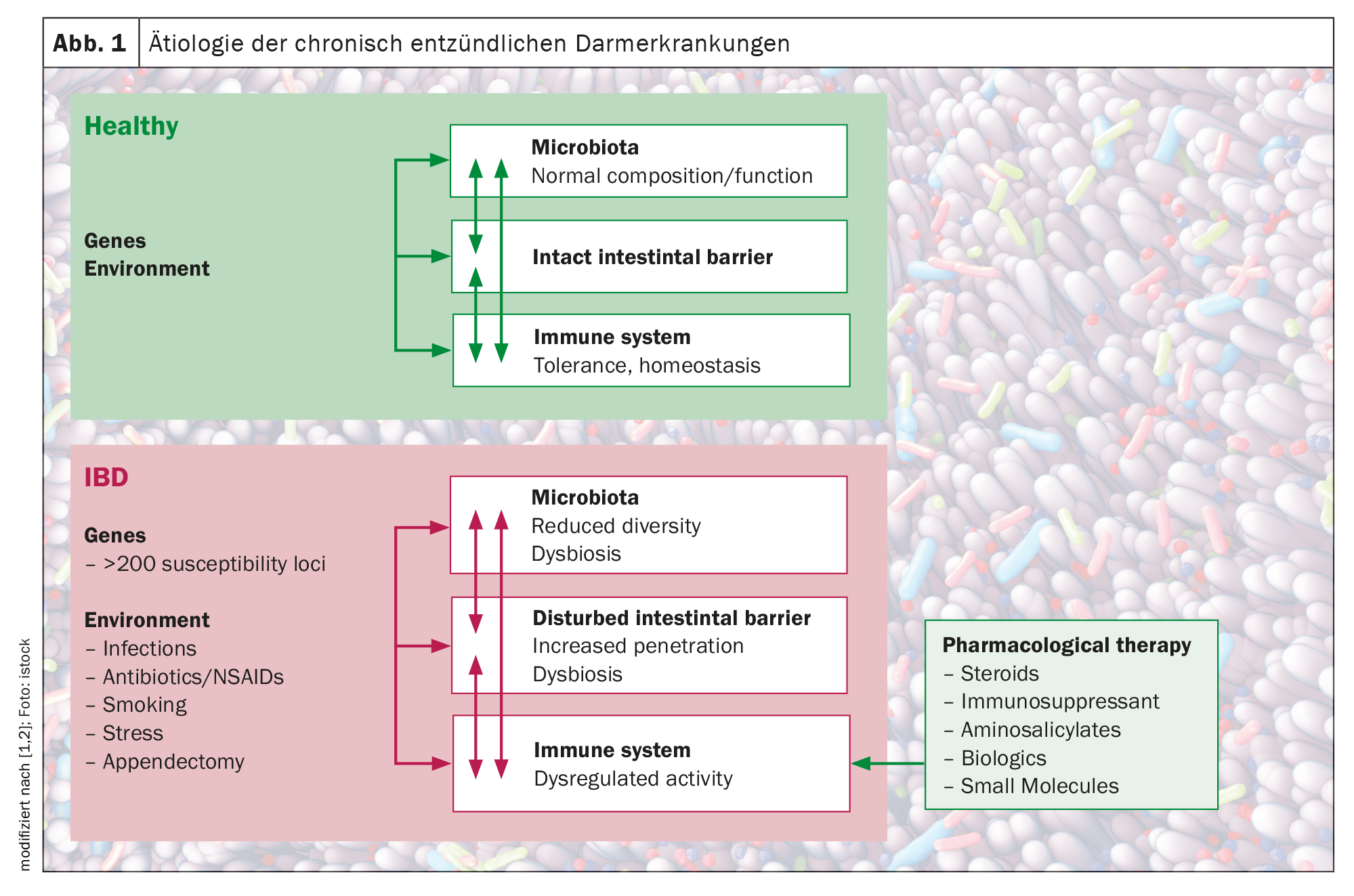
Gli studi hanno dimostrato che il trasporto alterato delle cellule immunitarie e le cellule immunitarie patogene sono fattori cruciali responsabili dell’infiammazione della mucosa e della distruzione dei tessuti nelle IBD. Una barriera intestinale difettosa e la disbiosi microbica portano a tale accumulo e all’attivazione locale delle cellule immunitarie, con il risultato di un ciclo di citochine pro-infiammatorie che scavalca i segnali anti-infiammatori e causa l’infiammazione cronica dell’intestino.
Le malattie infiammatorie intestinali (IBD), come la malattia di Crohn (CD) e la colite ulcerosa (UC), sono caratterizzate da un’attivazione incontrollata delle cellule immunitarie dell’intestino in un individuo geneticamente predisposto. Ad oggi, l’immunopatologia delle IBD non può essere spiegata completamente. Tuttavia, i singoli componenti che contribuiscono alla progressione di questo processo infiammatorio cronico, tra cui i fattori ambientali, i processi di migrazione alterati delle cellule immunitarie, così come i fattori genetici, microbici e immunologici, sono stati continuamente studiati. Dopo il contatto dell’organismo con un antigene, si verifica l’attivazione delle cellule presentanti l’antigene (APC) (risposta infiammatoria vs. tollerogenica). Le APC possono produrre mediatori come l’interleuchina 12, che porta all’attivazione, alla proliferazione e all’imprinting delle cellule T con un fenotipo intestinale, attraverso l’upregolazione di specifiche molecole di adesione. Una barriera intestinale difettosa e la disbiosi microbica portano all’accumulo e all’attivazione locale delle cellule immunitarie, con il risultato di un ciclo di citochine pro-infiammatorie che scavalca i segnali anti-infiammatori e causa l’infiammazione cronica dell’intestino. Gli studi di associazione genetica hanno identificato oltre 250 geni di suscettibilità per la malattia infiammatoria intestinale, rivelando aspetti fondamentali della biologia molecolare della malattia, compreso il ruolo dell’autofagia e della segnalazione e dello sviluppo delle cellule Th17.
Oltre alle influenze genetiche, tra cui i polimorfismi genetici dell’ospite in una serie di geni coinvolti nel riconoscimento e nell’elaborazione microbica, è stato riscontrato che anche i fattori ambientali come lo stile di vita, la dieta e i farmaci influiscono sull’equilibrio, spesso attraverso la loro influenza sulla composizione del microbiota intestinale. È ormai ampiamente accettato che l’IBD è il risultato di una “tempesta perfetta” di interazioni tra un microbiota disbiotico, un sistema immunitario anormale e influenze ambientali in un ospite suscettibile, spiega il Prof. Dr. Michael Scharl, Vice Direttore di IBD. Direttore della Clinica di Ricerca e Insegnamento di Gastroenterologia ed Epatologia, Ospedale Universitario di Zurigo (Fig. 1) [1,2].
Panoramica della struttura e della funzione del sistema immunitario associato all’intestino
Questa interruzione della barriera permette la traslocazione degli antigeni batterici presenti negli alimenti e in alcune regioni dal lume intestinale alla parete intestinale, dove poi incontrano la più grande raccolta di cellule immunitarie del corpo umano – il sistema immunitario muscolare, ha continuato Scharl. Il contatto con l’antigene è seguito dall’attivazione delle cellule presentanti l’antigene (APC) (risposta infiammatoria o tollerante). Le APC possono produrre mediatori come l’interleuchina 12, che porta all’attivazione, alla proliferazione e alla differenziazione delle cellule T con un fenotipo intestinale, attraverso la regolazione di specifiche molecole di adesione. Dopo il ricircolo, queste sottopopolazioni di cellule T possono successivamente migrare lungo gradienti chemiotattici verso l’intestino come tessuto bersaglio, dove interagiscono con molecole espresse dalle cellule endoteliali e avviano il processo di stravaso multistep dell’homing intestinale. Una volta giunte nel sito d’azione, le cellule T adattano la composizione delle loro molecole di superficie all’ambiente in cui si trovano, con conseguente ritenzione nel tessuto o, se non attivate, riciclo nel sangue e nella linfa. Se l’attivazione locale delle cellule T tramite la presentazione dell’antigene avviene nel tessuto intestinale, esse possono causare un potenziale danno massiccio nell’intestino infiammato [1,2].
Le risposte immunitarie deregolate causano l’IBD
Oltre ad un aumento del numero di cellule T, che sono particolarmente prevalenti nei pazienti con CD, sia i pazienti con CD che quelli con UC mostrano un aumento delle cosiddette cellule T helper di tipo 17 (cellule Th17), che producono la citochina caratteristica (interleuchina 17A). Inoltre, i pazienti con UC hanno anche un numero maggiore di cellule immunitarie di tipo 2 (cellule Th2), che producono interleuchina 5, per esempio. Le citochine tipiche delle cellule Th2 sono l’IL-4 e l’IL-13, che sono esplosioni di cellule immunitarie proinfiammatorie attivate, e queste risposte immunitarie proinfiammatorie sono contro-regolate da risposte immunitarie anti-infiammatorie mediate, ad esempio, da cellule T regolatorie (IL-10 e TGF-beta) o da cellule Th1. Queste cellule possono anche essere immunopatogene e presentare le seguenti citochine tipiche: IFN-gamma, TNF-alfa.
In particolare, lo squilibrio tra citochine pro-infiammatorie e anti-infiammatorie che si verifica nelle IBD ostacola la risoluzione dell’infiammazione e porta invece alla persistenza della malattia e alla distruzione dei tessuti. Le citochine svolgono un ruolo centrale nella modulazione del sistema immunitario intestinale. Sono prodotte da linfociti (soprattutto cellule T del fenotipo Th1 e Th2), monociti, macrofagi intestinali, granulociti, cellule epiteliali, cellule endoteliali e fibroblasti. Hanno funzioni proinfiammatorie [interleuchina-1 (IL-1), fattore di necrosi tumorale (TNF-alfa), IL-12] o antinfiammatorie [antagonista del recettore dell’interleuchina-1 (IL-1ra), IL-10, fattore di crescita trasformante β (TGFβ)]. Le concentrazioni mucosali e sistemiche di molte citochine pro- e anti-infiammatorie sono aumentate nelle IBD. Gli studi di associazione genome-wide hanno identificato diversi loci di suscettibilità per l’IBD che contengono geni che codificano citochine e proteine coinvolte nella segnalazione delle citochine. In particolare, è stato dimostrato che le mutazioni con perdita di funzione nei geni che codificano l’interleuchina-10 (IL-10) e il recettore dell’IL-10 sono associate a un’insorgenza molto precoce dell’IBD [3,4].
Cambiamenti nel microbiota intestinale a causa della terapia
Sono disponibili diversi farmaci per il trattamento dell’IBD. Spesso viene utilizzato un approccio terapeutico a stadi, passando da farmaci meno specifici, come l’acido 5-aminosalicilico, a farmaci più potenti, come i corticosteroidi, gli immunomodulatori e i biologici, a seconda della gravità dell’IBD. Oltre ai farmaci, l’unica altra opzione è la chirurgia (Fig. 2) [5].
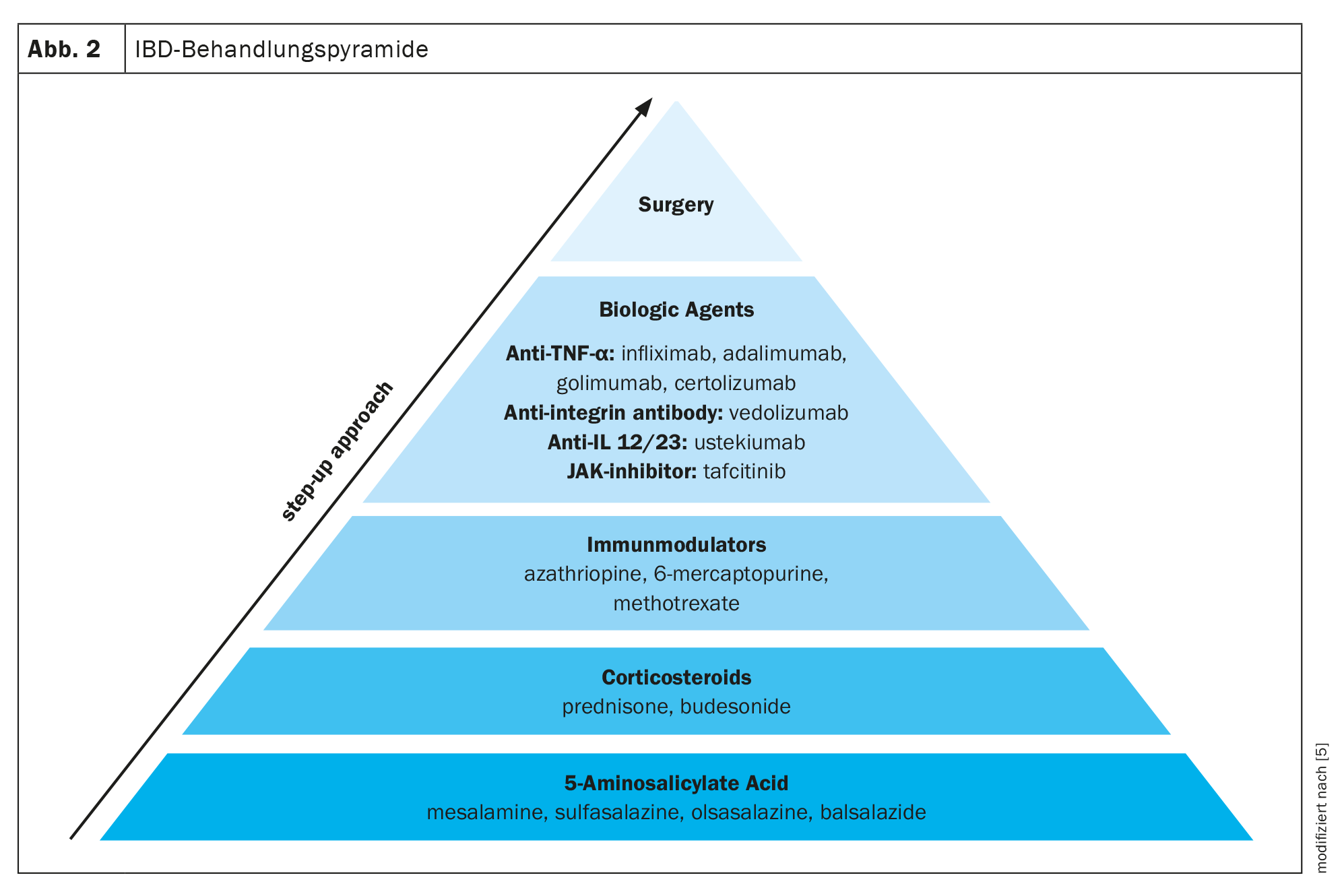
Uno sguardo dettagliato ai potenziali meccanismi d’azione delle terapie immunomodulanti attualmente disponibili mostra che esse si rivolgono a diversi potenziali bersagli del sistema immunitario mucosale, come le cellule immunitarie come i linfociti B, i macrofagi e i linfociti T, oltre a bersagli nell’area del traffico e della migrazione delle cellule T. Ustekinumab, ad esempio, blocca la differenziazione in cellule Th1 pro-infiammatorie; ozanimod inibisce la migrazione delle cellule T pro-infiammatorie dal linfonodo al linfatico drenante; Vedolizumab blocca specificamente la migrazione delle cellule T effettrici pro-infiammatorie dai vasi sanguigni ai tessuti intestinali; e gli anti-TNF, gli anti-IL12/23 e gli inibitori JAK bloccano la funzione o la trascrizione delle citochine per interrompere il ciclo infiammatorio [5].
Ustekinumab: differenziazione in cellule effettrici Th1 proinfiammatorie
Una visione dettagliata della differenziazione delle cellule T-helper nei linfonodi regionali mostra l’attivazione delle cellule di presentazione dell’antigene che producono l’interleuchina 12. Questo incontra le cellule T ingenue, che subiscono un’ulteriore differenziazione nelle cosiddette cellule Th1. Queste cellule Th1 polarizzate hanno recettori di homing come α4β7, che permettono loro di reinvadere il microbiota intestinale ed esprimere il recettore dell’interleuchina-12. Ustekinumab, che ha come bersaglio l’interleuchina 12, può sopprimere questa via di segnalazione e quindi inibire la polarizzazione delle cellule Th1. L’anticorpo monoclonale umano si lega specificamente alla subunità p40 dell’IL-12/23, impedendo all’IL-12 e all’IL-23 di legarsi ai loro complessi recettoriali sulla superficie cellulare, bloccando così i percorsi infiammatori T helper (Th) 1 (IL-12) e Th17 (IL-23). Ustekinumab è approvato in Svizzera sia per la CD che per l’UC e viene somministrato come infusione endovenosa durante l’induzione, con una dose di induzione di 6 mg/kg. Dopo una singola infusione, si passa alla terapia di mantenimento con somministrazione sottocutanea, 90 mg, q12w/q8w (revisione 1) [3,4].

Ozanimod: migrazione delle cellule T effettrici pro-infiammatorie dal linfonodo ai vasi linfatici drenanti.
Dopo il priming delle cellule T, emergono cellule effettrici polarizzate. Queste cellule lasciano il linfonodo regionale per raggiungere i vasi linfatici efferenti e il flusso sanguigno. Si tratta di un processo attivo che crea un gradiente chemiotattico mediato, almeno in parte, dalla molecola S1P. S1P si lega al recettore S1P sulle cellule T, questo percorso permette alle cellule di lasciare il linfonodo. Ozanimod, un agonista del recettore S1P, interferisce con questo punto finale del gradiente e impedisce l’uscita delle cellule T dal linfonodo regionale. Le cellule T colpite, altamente polarizzate, non possono più tornare in circolo. L’agonista del recettore S1P, precedentemente studiato nei pazienti con sclerosi multipla, è approvato in Svizzera per l’UC. Ozanimod viene somministrato per via orale in tre fasi, 0,23 mg il giorno 1-4 qd; 0,46 mg il giorno 5-7 qd; e 0,92 mg qd successivamente (Panoramica 2) [3,4].

Ozanimod è considerato una nuova opzione per i pazienti affetti da UC. Tuttavia, poiché si tratta di un agente biologico più recente nel trattamento delle IBD, sono necessari più dati reali oltre agli studi clinici per valutare dove l’agonista del recettore S1P si integra nella terapia delle IBD, spiega il Prof. Dr. Markus Neurath, Direttore della Clinica dell’Ospedale Universitario di Erlangen. In particolare, i reali effetti collaterali cardiovascolari e la necessità di un ECG, evidenziano la mancanza di esperienza nella pratica clinica di routine per il posizionamento di questo farmaco nei pazienti con UC, ha aggiunto Neurath.
Vedolizumab: migrazione delle cellule T effettrici pro-infiammatorie dai vasi sanguigni al tessuto intestinale
Vedolizumab è un anticorpo monoclonale umanizzato di immunoglobulina G1 (IgG1). Il suo meccanismo d’azione selettivo per l’intestino distingue il vedolizumab dagli attuali biologici disponibili per il trattamento dell’IBD, che si basano sull’immunosoppressione sistemica. L’anticorpo immunoglobulina G1 (IgG1) blocca in modo specifico l’integrina α4β7 sulla superficie della sottopopolazione di linfociti attivati che fluiscono nel flusso sanguigno e che sono predisposti all’homing nel tratto gastrointestinale. Questo blocco interrompe un meccanismo fisiopatologico essenziale delle IBD, che di solito permette ai linfociti di aderire all’endotelio del tratto gastrointestinale. Senza questa adesione, i linfociti non possono più migrare dal flusso sanguigno al tratto gastrointestinale infiammato, causando l’attenuazione dell’infiammazione localizzata e ponendo le basi per un controllo a lungo termine della malattia. Vedolizumab non interrompe il meccanismo di homing delle popolazioni di linfociti in altri tessuti, ad esempio nel sistema nervoso centrale (SNC), ma agisce come un farmaco diretto selettivamente alla parete intestinale attraverso un’immunosoppressione non sistemica. L’anticorpo immunoglobulina G1 (IgG1) è approvato per la CD e l’UC e viene somministrato per via endovenosa (300 mg alla settimana 0,2,6; poi 300 mg q8w) o sottocutanea (300 mg alla settimana 0,2; poi 108 mg q2w) (Panoramica 3) [3,4,6].

Effetti pro-infiammatori pleiotropici del TNF
Il TNF è un mediatore cruciale nel controllo dei processi infiammatori nell’intestino e viene utilizzato nella pratica clinica di routine da oltre 20 anni. Il TNF e i suoi recettori sono coinvolti in modo cruciale nella patogenesi delle IBD. Per esempio, sono stati riscontrati livelli maggiori della forma solubile di TNFR1 e TNFR2 sia nei pazienti con CD che con UC e la loro espressione è correlata all’attività della malattia.
Gli anticorpi anti-TNF bloccano sia la forma precursore transmembrana (mTNF) che la forma solubile (sTNF), riducendo così il milieu pro-infiammatorio nell’intestino, bloccando l’interazione tra il TNF e il recettore del TNF, e quindi bloccando vari tipi di cellule immunitarie pro-infiammatorie. Inoltre, il TNF provoca la morte delle cellule epiteliali. Gli anticorpi anti-TNF hanno quindi diversi meccanismi d’azione che possono essere utilizzati nella pratica clinica nei pazienti IBD sia con CD che con UC. Gli anticorpi anti-TNF collaudati per l’uso clinico di routine includono infliximab, adalimumab, golimumab e certoliizumab pegol, ciascuno con applicazioni diverse. Alcuni sono disponibili per la terapia endovenosa, altri sia per la terapia sottocutanea che per quella endovenosa e altri ancora solo per la somministrazione sottocutanea (revisione 4) [7].
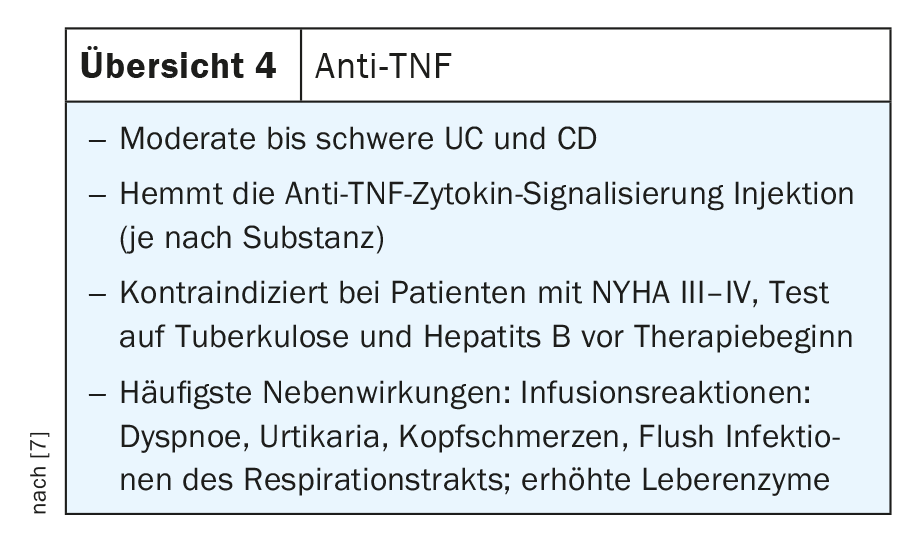
Secondo Neurath, i vari anticorpi anti-TNF si sono dimostrati validi nella pratica clinica e sono ancora oggi utilizzati in modo selettivo. Se per via endovenosa o sottocutanea, dipende in qualche modo dal contesto clinico. Se l’attività clinica è elevata o se il paziente è ricoverato in ospedale, la somministrazione per via endovenosa è certamente un buon modo per somministrare gli anticorpi anti-TNF, soprattutto nei pazienti con attività molto elevata che perdono molti anticorpi nelle feci. Neurath aggiunge che non è assolutamente necessario determinare le misurazioni del livello di valle o controllare lo stato degli anticorpi. Di solito questo viene fatto solo nei pazienti con una mancata risposta o una perdita secondaria di efficacia, oppure nei pazienti che non riescono a ottenere la risposta clinica primaria desiderata. In questo caso, ci sono diverse opzioni, aggiunge Neurath: passare a un altro agente o aggiungere un immunosoppressore come l’azatioprina per sopprimere le risposte delle cellule B e gli anticorpi anti-farmaco. Lo studio SONIC ha già dimostrato che la terapia combinata come azatioprina più infliximab è superiore alla monoterapia con uno solo dei due. Tuttavia, la terapia di combinazione dovrebbe dipendere dall’attività clinica. In alternativa, il paziente può essere monitorato clinicamente, ad esempio con ecografie per proteggere i livelli di CRP e attività cliniche per determinare se il paziente è in remissione clinica. Se si verificano problemi, la terapia può essere intensificata, ad esempio accorciando l’intervallo di infusione e aumentando la dose dell’agente attivo, oppure passare a un’altra classe di agenti biologici.
Attività delle cellule effettrici Th17 pro-infiammatorie
Ustekinumab è un anticorpo che blocca non solo l’IL 12, ma anche l’interleuchina 23. L’IL-23 porta all’attivazione e al mantenimento delle funzioni effettrici delle cellule Th17 proinfiammatorie nel tessuto intestinale. Ustekinumab riduce l’attività delle cellule Th17 pro-infiammatorie nel tessuto intestinale, bloccando l’interazione Il-23/Il-23R. Sulla base di queste scoperte, si sta attualmente cercando di sviluppare antagonisti selettivi contro l’interleuchina 23. Questi non sono destinati a colpire la subunità P40, come nel caso dell’ustekinumab, ma la subunità P19, che è unica dell’interleuchina 23 e non si trova nell’interleuchina 12. Alcuni agenti per questo sono già in fase di sperimentazione clinica e prima o poi saranno utilizzati nella pratica clinica, ad esempio risankizumab, mirikizumab, guselkumab e brazikumab. Inoltre, alcuni dati preliminari suggeriscono che i bloccanti P19 possono essere efficaci anche quando i bloccanti P40 non hanno funzionato in precedenza (revisione 5) [3,4].

Segnalazione delle citochine attraverso le vie di segnalazione JAK/STAT
La via di segnalazione della Janus chinasi e attivatore della trascrizione (JAK-STAT) svolge un ruolo importante nella trasmissione dei segnali dai recettori della membrana cellulare al nucleo. Molte citochine pro-infiammatorie inducono la trascrizione di geni effettori nella cellula bersaglio, attivando specifiche vie di segnalazione JAK/STAT. La famiglia JAK umana è composta da quattro JAK: JAK1, JAK2, JAK3 e TYK2. Il tofacitinib, un inibitore che ha come bersaglio principale JAK 1 e JAK 3, e in misura minore JAK 2, riduce la trascrizione dei geni di segnalazione ed effettori pro-infiammatori, bloccando l’attività della chinasi JAK. Tofacitinib è una piccola molecola che agisce su diverse citochine contemporaneamente ed è disponibile per via orale. Tuttavia, finora l’inibitore è stato approvato solo per l’UC, ma non per la CD (Panoramica 6) [8–10].

A causa della sua attività immunomodulante e del rischio di eventi cardiovascolari e tromboembolici, da quando è stato approvato il tofacitinib sono state imposte delle restrizioni sull’uso, che non lo rendono il farmaco di prima scelta, ha detto Neurath. In particolare, i pazienti anziani con malattie cardiovascolari e un rischio potenzialmente aumentato di eventi tromboembolici devono essere sottoposti a un attento esame prima di iniziare la terapia.
In Svizzera, attualmente è disponibile solo tofacitinib per l’inibizione di JAK. Tuttavia, grazie alla varietà di studi, gli inibitori della JAK sono sempre più approvati o in fase di sperimentazione clinica, per cui prima o poi sarà disponibile un’intera gamma di inibitori della JAK per la pratica clinica. In questo caso, i più piccoli cambiamenti nell’affinità delle molecole faranno una grande differenza clinica. Secondo Neurath, potrebbero esserci differenze rilevanti tra i vari inibitori di JAK-1, ad esempio, ma questo aspetto deve ancora essere indagato. La patogenesi chiarisce che questi agenti interferiscono con l’attivazione delle cellule immunitarie. Sarà interessante confrontare l’efficacia, ma soprattutto il profilo di sicurezza, aggiunge Neurath. Questo perché la sicurezza, in particolare, gioca un ruolo decisivo per la routine clinica, ma anche per i pazienti.
Opzioni future e terapie combinate per l’IBD
Secondo Neurath, sono disponibili i primi dati di uno studio di combinazione con bloccanti P19 più anticorpi anti-TNF e sono in corso ulteriori studi [11]. In sostanza, in questo caso è stata utilizzata una terapia combinata per indurre la remissione, quindi non si tratta di una terapia combinata a vita. Tuttavia, questa potrebbe essere un’opzione per i pazienti difficili da trattare. Vedolizimab in particolare sembra essere un partner interessante per le terapie di combinazione, perché ha un meccanismo d’azione molecolare molto diverso rispetto ad altri agenti, quindi è facile almeno postulare che ci possano essere sinergie, ha aggiunto Neurath. Inoltre, si tratta di un agente molto più sicuro e con un buon profilo di sicurezza, che lo rende una linea di base ideale per qualsiasi approccio combinato. Tuttavia, Neurath non vede un grande potenziale nella combinazione degli inibitori della JAK con gli agenti biologici.
Messaggi da portare a casa
- Le terapie immunomodulanti si rivolgono a diversi potenziali bersagli del sistema immunitario della mucosa.
- Ustekinumab blocca la differenziazione in cellule Th1 (tramite l’IL-12), nonché la citochina IL-23.
- Ozanimod inibisce la migrazione delle cellule T pro-infiammatorie dal linfonodo ai vasi linfatici drenanti.
- Vedolizumab blocca in modo specifico la migrazione delle cellule T effettrici pro-infiammatorie dai vasi sanguigni al tessuto intestinale.
- Gli anti-TNF, gli anti-IL12/23 e gli inibitori JAK bloccano la funzione delle citochine per arrestare l’infiammazione.

Letteratura:
- Neurath MF: Puntare i circuiti e il traffico delle cellule immunitarie nella malattia infiammatoria intestinale. Nat Immunol 2019; doi: 10.1038/s41590-019-0415-0.
- de Lange KM, et al: Lo studio di associazione genome-wide implica l’attivazione immunitaria di più geni integrina nella malattia infiammatoria intestinale. Nat Genet 2017; doi: 10.1038/ng.3760.
- Neurath MF: Citochine nella malattia infiammatoria intestinale. Nat Rev Immunol 2014; doi: 10.1038/nri3661.
- Neurath MF: Obiettivi terapeutici attuali ed emergenti per le IBD. Nat Rev Gastroenterol Hepatol 2017; doi: 10.1038/nrgastro.2016.208.
- Wu N, et al: La malattia infiammatoria intestinale e il microbiota intestinale. Proc Nutr Soc 2021; doi: 10.1017/S002966512100197X.
- Denucci CC, et al.: Funzione dell’integrina nell’homing delle cellule T verso siti linfoidi e non linfoidi: arrivare e rimanere. Crit Rev Immunol 2009; doi: 10.1615/critrevimmunol.v29.i2.10.
- Billmeier U, et al.: Meccanismo d’azione molecolare degli anticorpi contro il fattore di necrosi tumorale nelle malattie infiammatorie intestinali. World J Gastroenterol 2016; doi: 10.3748/wjg.v22.i42.9300.
- Vetter M, Neurath M: Terapie orali mirate emergenti nelle malattie infiammatorie intestinali: opportunità e sfide. Therap Adv Gastroenterol 2017; doi: 10.1177/1756283X17727388.
- Seif F, et al.: Il ruolo della via di segnalazione JAK-STAT e dei suoi regolatori nel destino delle cellule T helper. Cell Commun Signal 2017; doi: 10.1186/s12964-017-0177-y.
- Danese S, et al: Selettività JAK per il trattamento delle malattie infiammatorie intestinali: ha importanza clinica? Gut 2019; doi: 10.1136/gutjnl-2019-318448.
- Feagan BG, et al: Terapia combinata guselkumab più golimumab rispetto alla monoterapia con guselkumab o golimumab in pazienti con colite ulcerosa (VEGA): uno studio randomizzato, in doppio cieco, controllato, di fase 2, proof-of-concept. Pubblicato :01 febbraio 2023. DOI: https://doi.org/10.1016/S2468-1253(22)00427-7
HAUSARZT PRAXIS 2023: 18(6): 6–11










