Oltre all’ematomatosi, la fibrosi cistica (FC) è la malattia genetica più comune e la più comune malattia genetica con coinvolgimento polmonare. Nei pazienti adulti, i sintomi sono vari e spesso non si distinguono a prima vista da altri quadri clinici. Un’anamnesi accurata e gli esami giusti aiutano a rintracciare la malattia.
La FC non è più solo una malattia infantile – il 51,2% delle persone iscritte nei registri è in età adulta [1]. In Svizzera, circa 1000 persone ne sono affette, ha ricordato il dottor Macé Schuurmans, Clinica di Pneumologia, Ospedale Universitario di Zurigo [2]. La FC si eredita in modo autosomico recessivo, per cui esistono circa 2000 costellazioni diverse di cambiamenti ereditari. Il gene affetto codifica una proteina multifunzionale, la CFTR (Cystic Fibrosis Transmembrance Regulator), che serve come canale del cloruro. Esistono mutazioni gravi (classe I-III) che sono associate all’insufficienza pancreatica. La mutazione più comune che abbiamo in Svizzera è mostrata qui e riguarda una delezione nel sito 508 (p.Phe508del) per la fenilalanina in questo gene. L’insufficienza pancreatica si verifica perché il paziente ha da un lato il diabete, ma dall’altro anche un problema digestivo perché gli enzimi digestivi non sono funzionali.
Le terapie con modulatori CFTR sono disponibili da circa 5 anni (ivacaftor, tezacaftor, lumacaftor, elexacaftor). Le sostanze, singolarmente o in combinazione, influenzano la funzione del prodotto genico sia qualitativamente che quantitativamente. Questa innovazione ha portato a un importante progresso nella terapia della FC, almeno per i pazienti con le mutazioni molto comuni.

Diagnosi
Dal 2011, esiste uno screening neonatale, che viene effettuato con un test del tripsinogeno immunoreattivo (IRT) seguito da uno screening del DNA. Di solito al 4° giorno di vita, vengono prelevate alcune gocce di sangue dal tallone del bambino e vengono poste su un cartoncino di carta da filtro per essere analizzate nel laboratorio di screening. Questo esame copre il 98% di tutte le malattie della FC.
I vantaggi di eseguire il test il più precocemente possibile sono evidenti: “Sappiamo da diversi Paesi che hanno introdotto lo screening precocemente che il percorso verso la diagnosi è significativamente più breve”, ha detto il dottor Schuurmans. In questo modo, i genitori possono essere consigliati sulle future gravidanze, tra le altre cose. La malattia si manifesta in modo meno grave se viene trattata correttamente e precocemente, e la funzione polmonare non si riduce altrettanto rapidamente.
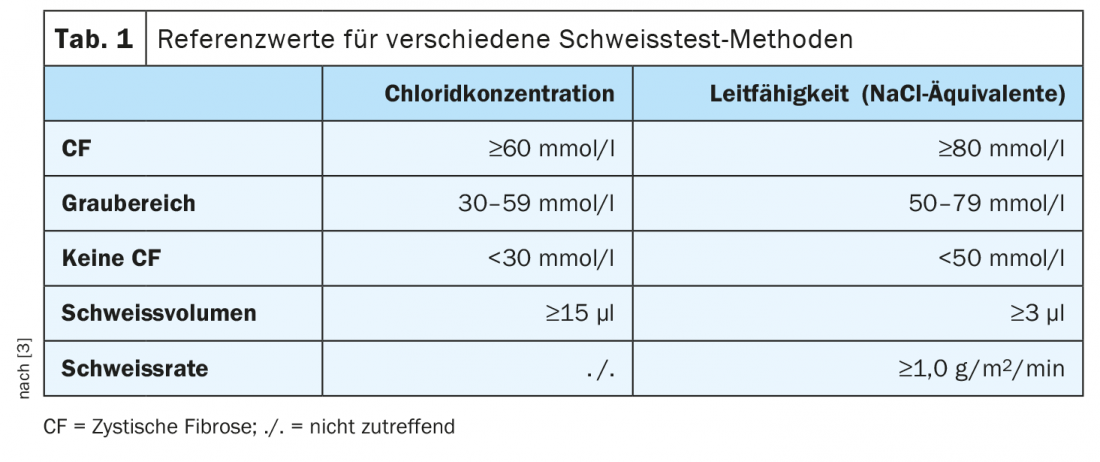
Prima del 2011, questo screening non era disponibile – tutti gli individui nati prima di allora, così come gli adulti che presentano una diagnosi tardiva, tendono ad avere forme più lievi che non sono necessariamente così chiare nell’infanzia. Il primo passo più importante quando si sospetta la FC in questo gruppo di pazienti è il test del sudore, che è considerato il gold standard per i bambini nati prima del 2011 e per gli adulti. Il test determina sia il contenuto di cloruro che la conduttività. In un caso tipico, il contenuto di cloruro è superiore a 60 mmol/l, una gamma grigia esiste tra 30 e 59 mmol/l. Se non è sicuro della diagnosi, può usare la conduttività come aiuto. Se il valore scende al di sotto di 30 mmol/l, invece, è chiaro che la FC non è presente (Tab. 1) [3].
Quando pensare alla FC
Negli adulti, si dovrebbe pensare alla FC se si presentano alcuni sintomi e chiedere anche l’anamnesi familiare, consiglia il dottor Schuurmans. I sintomi includono:
- Infezioni respiratorie ricorrenti, soprattutto con alcuni “germi problematici” come lo stafilococco. aureus, Pseudomonas aeruginosa, Burkholderia cepacia complex o anche micobatteri atipici (NTM).
- Asma bronchiale atipica, cioè con tosse cronica produttiva che non risponde alla terapia standard.
- Bronchiectasie che si manifestano prima dei 40 anni. “Questo può accadere anche in seguito”, dice l’esperto. “Ma prima dei 40 anni, l’esclusione del FC è obbligatoria”.
- Poliposi nasale, sinusite grave
- Infertilità negli uomini (a volte può essere l’unico segno nelle fasi iniziali)
- Disturbo elettrolitico (ad esempio, iponatriemia, disidratazione)
- Pancreatite acuta
- Malattia epatica inspiegabile
Se si sospetta la FC negli adulti, si raccomanda innanzitutto il test del sudore o il rinvio a un consulto sulla FC per adulti. Il test del sudore viene classicamente eseguito nell’ospedale pediatrico. La genetica è solo il secondo passo, ed è obbligatorio che si svolga un colloquio di consulenza da parte di medici adeguatamente formati.
Se la fibrosi cistica non viene scoperta prima dei 30 anni, la sopravvivenza è di conseguenza più lunga. Tuttavia, se un paziente viene scoperto precocemente e la sua funzione polmonare è molto buona, si hanno molti più anni prima che venga effettuato un trapianto o che il paziente muoia, ha riassunto il dottor Schuurmans.
Congresso: FomF WebUp Pneumologia
Fonti:
- Zolin A, Orenti A, Naehrlich L, et al: Relazione annuale ECFSPR 2018.
- FomF WebUp Pneumologia, 7.12.2020.
- Jung A: Schweiz Med Forum 2017; 17(24): 514-522.
HAUSARZT PRAXIS 2021; 16(2): 37 (pubblicato il 19.2.21, prima della stampa).
InFo PNEUMOLOGIA & ALLERGOLOGIA 2021; 3(1): 36