La leucemia linfoblastica acuta è il tumore infantile più comune. Viene trattata in modo adeguato al rischio e nella maggior parte dei casi è curabile. Nuovi farmaci innovativi, come le immunoterapie, sono in fase di sperimentazione clinica.
Con una quota di circa il 30% e 3,3 nuovi casi per 100.000 abitanti di età inferiore ai 15 anni, la leucemia linfoblastica acuta (ALL) è il tumore infantile più comune. Il picco di età è di circa cinque anni. In Svizzera, ogni anno vengono diagnosticati circa 50-60 bambini con ALL. Il sottotipo immunologicamente più comune nell’infanzia è l’ALL a precursore B, che si sviluppa da cellule immature della serie B del sistema linfatico. TUTTA la linfopoiesi T si verifica meno frequentemente. Una forma particolare è la leucemia a cellule B mature, che si basa su una trasformazione maligna delle cellule B mature ed è intesa come una manifestazione leucemica del linfoma di Burkitt. L’ALL è una malattia eterogenea caratterizzata dalla proliferazione incontrollata di cellule progenitrici linfoidi nel midollo osseo e nel sangue periferico [1]. Oggi si ritiene che sia una malattia che spesso presenta grandi somiglianze morfologiche, ma che può avere sottogeneri citogenetici o genetici molecolari molto eterogenei [2], il che si correla anche con una risposta clinica eterogenea al trattamento. Con le moderne tecniche di sequenziamento, è possibile dimostrare l’enorme eterogeneità clonale di questa malattia.
Cause
La causa dello sviluppo della leucemia non è ancora chiara. I fattori noti, ma che si verificano raramente, sono le radiazioni ionizzanti e le sindromi congenite. Tuttavia, questo spiega meno del 10% di tutte le malattie. I bambini con sindrome di Down hanno un rischio circa 20 volte superiore di sviluppare una leucemia (ALL o leucemia mieloblastica acuta) entro i primi cinque anni di vita, rispetto ai bambini sani non affetti. Tuttavia, una mieloproliferazione transitoria si verifica ancora più frequentemente (nel 3-10%) in questi bambini in età neonatale, che occasionalmente può trasformarsi in seguito in leucemia. Altre rare alterazioni congenite che comportano un aumento del rischio di leucemia sono l’atassia teleangiectatica, la sindrome di Fanconi e altre sindromi associate a un’immunodeficienza o a una maggiore fragilità cromosomica. Il fatto che l’ALL si manifesti più frequentemente tra il secondo e il quinto anno di vita, che la malattia sia più comune nei Paesi industrializzati e l’osservazione che in passato si sono verificati ripetutamente dei raggruppamenti, soprattutto nelle regioni di nuovi agglomerati, hanno portato a varie ipotesi associate alle infezioni per lo sviluppo della leucemia [3,4].
Sintomi
La proliferazione dei blasti leucemici nel midollo osseo porta a uno spostamento della normale ematopoiesi, il che spiega i sintomi più comuni, come il pallore e l’affaticamento dovuti all’anemia o la tendenza al sanguinamento dovuta alla trombocitopenia. Le infiltrazioni causano spesso dolore osseo diffuso e artropatie alternate, che occasionalmente si manifestano nei bambini piccoli come riluttanza a muoversi o addirittura rifiuto di camminare. Inoltre, possono verificarsi gonfiore dei linfonodi e organomegalia.
Diagnostica
Nel sangue, spesso si riscontrano alterazioni in almeno due serie di cellule ematiche, più frequentemente trombocitopenia con contemporanea anemia. La conta leucocitaria può essere normale, diminuita o aumentata. La morfologia dell’emocromo fornisce importanti indizi diagnostici; la diagnosi finale viene fatta con un’aspirazione del midollo osseo. Oltre all’esame della morfologia, l’immunofenotipo dei blasti leucemici viene determinato mediante la citometria a flusso (FACS) e viene effettuata un’analisi cromosomica. L’immunofenotipizzazione consente di determinare la fase di sviluppo del clone cellulare corrispondente. Il sottotipo di leucemia più comune nei bambini, la cosiddetta “common-ALL”, è caratterizzata dall’espressione dei marcatori delle cellule B CD10 e CD19. L’espressione di antigeni mieloidi, di solito privi di significato prognostico, può essere rilevata fino alla metà dei casi di ALL. Al giorno d’oggi, gli esami citogenetici e di genetica molecolare stanno diventando sempre più importanti. È importante riconoscere i sottogruppi più importanti, in quanto hanno implicazioni terapeutiche. Da un lato, si cercano cambiamenti cromosomici numerici come l’iperdiploidia o l’ipodiploidia, nonché cambiamenti strutturali come traslocazioni, ad esempio t(12;21) (fusione dei geni ETV6/RUNX1) o t(9;22) (fusione di BCR/ABL1), riarrangiamenti MLL (MLL 11q23) e altri cambiamenti.
Classicamente, il rilevamento di questi cambiamenti avviene con la citogenetica convenzionale (G-banding) e/o l’ibridazione fluorescente in situ (FISH) nelle cellule leucemiche. Negli ultimi anni, la misurazione della malattia minima residua (MRD) dal midollo osseo si è affermata come parte della diagnostica di follow-up per valutare la risposta della leucemia al trattamento. La risposta alla terapia è emersa come uno dei parametri prognostici più importanti. Oggi, per la diagnosi della progressione si utilizzano essenzialmente due metodi che si completano a vicenda nella pratica clinica quotidiana. Il metodo più sensibile è il monitoraggio dei riarrangiamenti dei recettori delle immunoglobuline e delle cellule T. Inizialmente, si cercano i riarrangiamenti clonali specifici della leucemia, che vengono seguiti con la PCR quantitativa in momenti specifici della terapia. Il limite di rilevazione raggiunto con questo metodo è di circa una cellula leucemica ogni 100.000 cellule normali del midollo osseo. Una tecnica per la misurazione della MRD, che è meno sensibile di circa un livello logico, si basa sul monitoraggio dell’immunofenotipo associato alla leucemia mediante FACS. Si può raggiungere una sensibilità dello 0,001% [5].
Trattamento
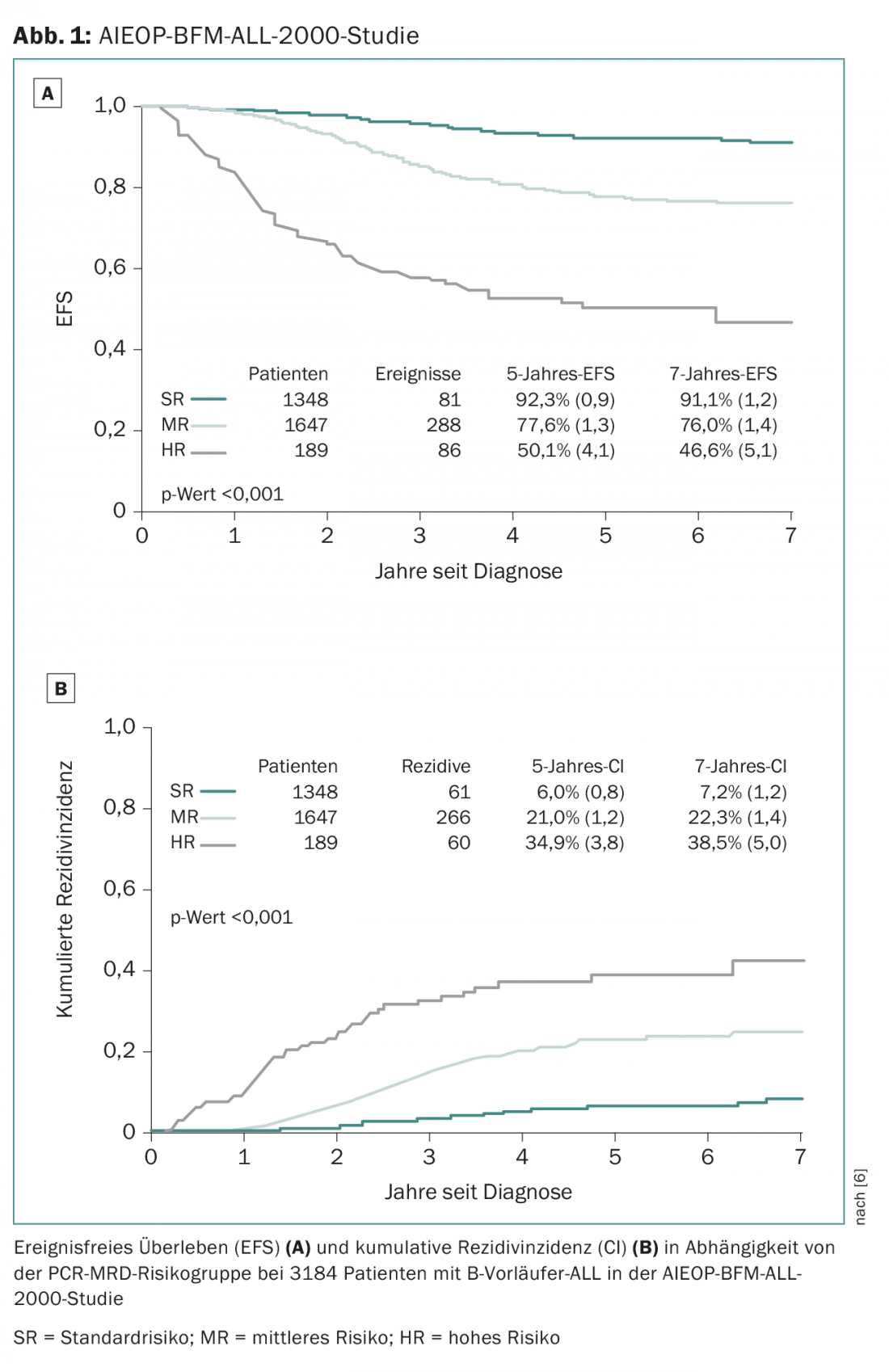
Negli anni ’70, meno del 30% dei bambini sopravviveva alla malattia, mentre oggi quasi l’85% dei pazienti può essere curato a lungo termine (Fig. 1) . I progressi nei tassi di sopravvivenza negli ultimi 10-15 anni sono stati raggiunti soprattutto grazie a una migliore stratificazione del rischio e al conseguente trattamento adattato al rischio. L’attuale terapia di prima linea di ALL consiste essenzialmente in una combinazione di corticosteroidi, deplezione di aminoacidi o substrati (asparaginasi, metotrexato), sostanze alchilanti, antimetaboliti, bloccanti classici della metafase e antracicline [6]. Le sostanze più recenti, le cosiddette terapie “mirate”, sono state finora utilizzate solo in misura molto limitata nel trattamento dell’ALL pediatrica, con alcune eccezioni come gli inibitori della tirosin-chinasi nell’ALL positiva al cromosoma Philadelphia. Recentemente, sono stati identificati nuovi sottogruppi rari di ALL a precursore B, i cosiddetti “Philadelphia like” (o “BCR-ABL like”), che da un lato mostrano un rischio significativamente aumentato di recidiva, ma dall’altro potrebbero essere potenziali candidati per terapie mirate [7]. Per la maggior parte dei nuovi criteri genetici ad alto rischio, tuttavia, si è potuto anche dimostrare che la loro influenza varia, a seconda della risposta terapeutica misurabile.
La maggior parte dei centri di oncologia pediatrica svizzeri tratta i propri pazienti nell’ambito degli studi del gruppo di studio ALL-BFM, un’associazione di centri di oncologia pediatrica tedeschi, austriaci e svizzeri, che ha contribuito in modo significativo ai miglioramenti terapeutici nell’ALL in numerosi studi terapeutici randomizzati su larga scala dal 1976. Dopo che la maggior parte dei progressi nei precedenti studi ALL-BFM è stata ottenuta grazie agli aggiustamenti nella classificazione dei gruppi di rischio e all’individualizzazione del trattamento, lo studio di follow-up AIEOP-BFM ALL 2017, che è ora in fase di pianificazione, sarà il primo ad utilizzare nuovi farmaci promettenti. La cosiddetta “spina dorsale” del trattamento è rappresentata dai farmaci classici del trattamento di ALL sopra menzionati. Inoltre, i nuovi farmaci innovativi vengono testati in modo casuale per verificarne i potenziali benefici. Il previsto studio AIEOP-BFM-2017 definirà nuovi gruppi citogenetici ad alto rischio (panoramica 1) che avranno accesso a nuovi approcci terapeutici innovativi. Uno di questi nuovi sottogruppi è definito dalla presenza di una delezione di IKZF1 in combinazione con una delezione di CDKN2A, CDKN2B, PAX5 o PAR1 in assenza di una delezione ERG e viene definito IKZF1plus. In studi precedenti, circa il 10-15% dei pazienti poteva essere identificato come IKZF1plus e questi avevano un tasso di recidiva significativamente più alto rispetto ai pazienti IKZF1plus-negativi [8]. Uno di questi nuovi farmaci, che sarà utilizzato in un piccolo gruppo di gruppi ad alto rischio con una prognosi particolarmente sfavorevole, è il blinatumomab, un anticorpo bispecifico delle cellule T (BiTE) che ha come bersaglio simultaneo il recettore CD3 delle cellule T e la proteina di superficie delle cellule B, il CD19 [9]. Blinatumomab è destinato a combinare due effetti potenziali: Riduzione delle tossicità acute e a lungo termine, risparmiando la chemioterapia convenzionale e una terapia più efficace dei pazienti che finora hanno risposto in modo insoddisfacente solo alla terapia ad alto rischio.

Un altro nuovo farmaco con una nuova modalità d’azione nella terapia di prima linea di ALL è l’inibitore del proteasoma bortezomib. Poiché i precedenti tentativi di intensificazione tardiva della terapia nei pazienti ad alto rischio hanno avuto scarso successo, e a causa delle tossicità già elevate delle terapie ad alto rischio (HR), il bortezomib sarà randomizzato ai pazienti HR nella fase iniziale post-reinduzione nel prossimo studio.
Sistema nervoso centrale (SNC)
Al giorno d’oggi, la prevenzione di una ricaduta a livello del SNC viene effettuata principalmente con i farmaci, da un lato con iniezioni intratecali di metotrexato, dall’altro con la somministrazione di farmaci citostatici ad azione sistemica che infiltrano il cervello (ad esempio, metotrexato ad alte dosi). Questo ha permesso di limitare la precedente radioterapia del SNC, che ha portato a una drastica riduzione delle recidive del SNC ma è stata associata a effetti tardivi non trascurabili, a situazioni di rischio molto particolari [10,11].
Trapianto di cellule staminali
Dopo che i risultati della terapia primaria e i protocolli di ricaduta delle leucemie sono migliorati in modo significativo nel tempo, questo ha portato anche a un continuo adeguamento dell’indicazione per le terapie ad alto dosaggio con reinfusione di cellule staminali. L’attuale indicazione del trapianto di cellule staminali (SCT) come parte della terapia primaria è riservata ad alcuni sottogruppi citogenetici prognosticamente sfavorevoli, come t(9;22), ipodiploidie con meno di 44 cromosomi nei blasti, e IKZF1plus in combinazione con una risposta insufficiente alla terapia (MRD) nel tempo [12]. L’esperienza del gruppo BFM ha dimostrato che il successo del trattamento delle ricadute dipende dal momento in cui si verifica la ricaduta, dal modello della leucemia e dal sottotipo di leucemia [13]. Tuttavia, è stato anche dimostrato che la risposta alla terapia dopo l’induzione della terapia rinnovata e quindi la dinamica del declino della malattia minima residua è di particolare importanza prognostica e ulteriori elementi terapeutici, come l’uso della SCT, possono essere allineati di conseguenza [14].
Nuove terapie
A parte alcune eccezioni (clofarabina, nelarabina, imatinib), non ci sono state nuove approvazioni per l’ALL pediatrica negli ultimi 10-15 anni. Tuttavia, attualmente ci sono diversi approcci terapeutici interessanti in fase di sperimentazione clinica negli studi di fase I-III. Oltre al già citato blinatumomab, questi includono anche le cellule T chimeriche con recettore dell’antigene (CAR), con le quali sono già state trattate con successo le recidive di ALL CD19-positive. Si tratta anche di un’immunoterapia che sfrutta il potenziale dei linfociti T citotossici autologhi per riconoscere e distruggere le cellule della linea cellulare B. Altre promettenti immunoterapie, alcune delle quali accoppiate a citostatici, sono attualmente in fase di sperimentazione I/II. Oltre alle immunoterapie, anche le terapie mirate, dopo una precedente sperimentazione in vitro in modelli di xenotrapianto o di linea cellulare, o gli inibitori specifici contro i geni di fusione individuati citogeneticamente, rappresentano opzioni terapeutiche interessanti e promettenti.
Messaggi da portare a casa
- La leucemia linfoblastica acuta, il tumore infantile più comune, viene trattata in modo adeguato al rischio ed è curabile nella maggior parte dei casi.
- La determinazione della malattia minima residua dopo l’induzione della terapia è uno dei fattori prognostici più importanti, insieme ai marcatori biologici come il sottotipo leucemico e le modifiche citogenetiche e genetiche molecolari nei blasti leucemici.
- Gli sviluppi attuali mirano a un trattamento più efficace e mirato dei sottotipi di leucemia precedentemente resistenti, nonché a una riduzione della tossicità della terapia.
- Nuovi farmaci innovativi, come le immunoterapie e gli approcci terapeutici individualizzati, sono in fase di sperimentazione clinica.
Letteratura:
- Jabbour E, et al: Nuove intuizioni sulla fisiopatologia e sulla terapia della leucemia linfoblastica acuta dell’adulto. Cancro 2015; 121(15): 2517-2528.
- Pui CH, et al: Biologia, stratificazione del rischio e terapia delle leucemie acute pediatriche: un aggiornamento. J Clin Oncol 2011; 29(5): 551-565.
- Kinlen L, et al: Infezioni e fattori immunitari nel cancro: il ruolo dell’epidemiologia. Oncogene 2004; 23: 60-75.
- Greaves M, et al: Infezione, risposte immunitarie ed eziologia della leucemia infantile. Nat Rev Cancer 2006; 6(3): 193-203.
- Campano D, et al: Monitoraggio della malattia minima residua nella leucemia linfoblastica acuta infantile. Curr Opin Hematol 2012; 19: 313-318.
- Conter V, et al: La risposta molecolare al trattamento ridefinisce tutti i fattori prognostici nei bambini e negli adolescenti con leucemia linfoblastica acuta precursore delle cellule B: risultati in 3184 pazienti dello studio AIEOP-BFM ALL 2000. Sangue 2010; 115(16): 3206-3214.
- Loh ML, et al: Sequenziamento del chinoma della tirosina nella leucemia linfoblastica acuta pediatrica: un rapporto del Children’s Oncology Group TARGET Project. Sangue 2013; 121(3): 485-488.
- Hinze L, et al: Impatto prognostico delle delezioni IKZF1 in associazione con gli impulsi di vincristina-desametasone durante il trattamento di mantenimento della leucemia linfoblastica acuta infantile nello studio ALL-BFM 95. Leucemia 2017; 31: 1840-1842.
- Brentjens RJ, et al.: Sicurezza e persistenza di cellule T autologhe trasferite adottivamente con target CD19 in pazienti con leucemie a cellule B recidivate o refrattarie alla chemioterapia. Sangue 2011; 118(18): 4817-4828.
- Möricke A, et al.: La terapia adattata al rischio della leucemia linfoblastica acuta può ridurre l’onere del trattamento e migliorare la sopravvivenza: risultati del trattamento di 2169 pazienti pediatrici e adolescenti non selezionati, arruolati nello studio ALL-BFM 95. Blood 2008; 111(9): 4477-4489.
- Kamps WA, et al: Trattamento orientato alla BFM per i bambini con leucemia linfoblastica acuta senza irradiazione cranica e riduzione del trattamento per i pazienti a rischio standard: risultati del protocollo DCLSG ALL-8 (1991-1996). Leucemia 2002; 16(6): 1099-1111.
- Balduzzi A, et al: Chemioterapia versus trapianto allogenico per la leucemia linfoblastica acuta infantile ad altissimo rischio nella prima remissione completa: confronto mediante randomizzazione genetica in uno studio prospettico internazionale. Lancet 2005; 366: 635-642.
- Tallen G, et al: Esito a lungo termine nei bambini con leucemia linfoblastica acuta recidivata dopo la stratificazione del punto di tempo e del sito di recidiva e la chemioterapia multifarmaco a breve durata intensificata: risultati dello studio ALL-REZ BFM 90. J Clin Oncol 2010; 28: 2339-2347.
- Eckert C, et al: La malattia minima residua dopo l’induzione è il più forte predittore di prognosi nella leucemia linfoblastica acuta recidivata a rischio intermedio – Risultati a lungo termine dello studio ALL-REZ BFM P95/96. Eur J Cancer 2013 Apr; 49(6): 1346-1355.
InFo ONCOLOGIA & EMATOLOGIA 2017; 5(5): 30-33