Il trattamento chirurgico dei pazienti con malattie ereditarie del tessuto connettivo mira a prevenire la dissezione aortica. Perché questo è responsabile dell’alto tasso di mortalità della popolazione di pazienti. Quando viene data l’indicazione? E quali sono le opzioni di terapia farmacologica?
Le malattie ereditarie del tessuto connettivo con manifestazioni vascolari sono difficilmente presenti nella coscienza del medico di base. Per molto tempo, la sindrome di Marfan è stata l’unica diagnosi differenziale in questa direzione nei pazienti giovani con un evento aortico toracico. Nell’ultimo decennio sono stati identificati centinaia di geni associati a forme sindromiche e non sindromiche di malattia aortica [1].
Inoltre, quasi la metà dei pazienti viene diagnosticata solo nel contesto di una complicazione vascolare. Mentre gli aneurismi vengono solitamente rilevati durante un esame di routine, la dissezione aortica è una delle emergenze chirurgiche associate a un’elevata morbilità e mortalità.
La sindrome di Marfan è un disturbo autosomico dominante con un’incidenza di circa 1:5000 nati vivi. Le tipiche manifestazioni oculari, scheletriche e cardiache sono causate da mutazioni nel gene della fibrillina-1, che portano alla sovraattivazione della via di segnalazione del TGFβ [2].
Bart Loeys e Hal Dietz hanno identificato dieci anni fa una sottopopolazione di pazienti che si distinguevano per un’ugola divisa, un’ampia distanza interoculare e vasi tortuosi. Nel frattempo, sono stati identificati diversi geni che portano a un fenotipo del gruppo della sindrome di Loeys-Dietz. L’identificazione di questi pazienti è importante perché le dissezioni spesso si verificano a diametri aortici che in precedenza non erano considerati indicazioni per la sostituzione aortica profilattica [3].
La rara sindrome vascolare di Ehlers-Danlos è causata da mutazioni nel gene che codifica per il collagene III. Questi pazienti si distinguono per l’alto tasso di dissezione e rottura senza precedente formazione di aneurisma. Questo rende la cura di questi pazienti molto difficile. La sopravvivenza mediana è di 48 anni se non trattata, con i primi eventi che di solito si verificano nella terza o quarta decade di vita [4].
Opzioni di trattamento chirurgico
L’obiettivo del trattamento chirurgico dei pazienti con malattie ereditarie del tessuto connettivo è quello di prevenire la dissezione aortica dovuta alla dilatazione dell’aorta, poiché questa è responsabile dell’elevata mortalità in questa popolazione di pazienti. Le linee guida della Società Europea di Cardiologia (ESC) del 2014 fanno una chiara distinzione nell’indicazione per la sostituzione aortica elettiva tra i pazienti Marfan o Loeys-Dietz e i pazienti senza malattia genetica sottostante [5]. La difficoltà sta nella demarcazione. Nei pazienti giovani, si deve presumere la presenza di una componente genetica. Nei pazienti senza malattia del tessuto connettivo, la sostituzione aortica profilattica è raccomandata quando il diametro della radice aortica o dell’aorta ascendente è di 55 mm. I pazienti Marfan, invece, dovrebbero essere operati a 50 mm, e in caso di fattori di rischio esistenti (come dissezioni in famiglia) anche a partire da 45 mm. Nei pazienti Loeys-Dietz, la sostituzione elettiva è consigliata a 42-45 mm. Di norma, viene sostituita soprattutto la radice aortica. Se possibile, si cerca di preservare la valvola del paziente. Se questo non è possibile a causa di un danno alla valvola, viene impiantato un condotto portante la valvola.
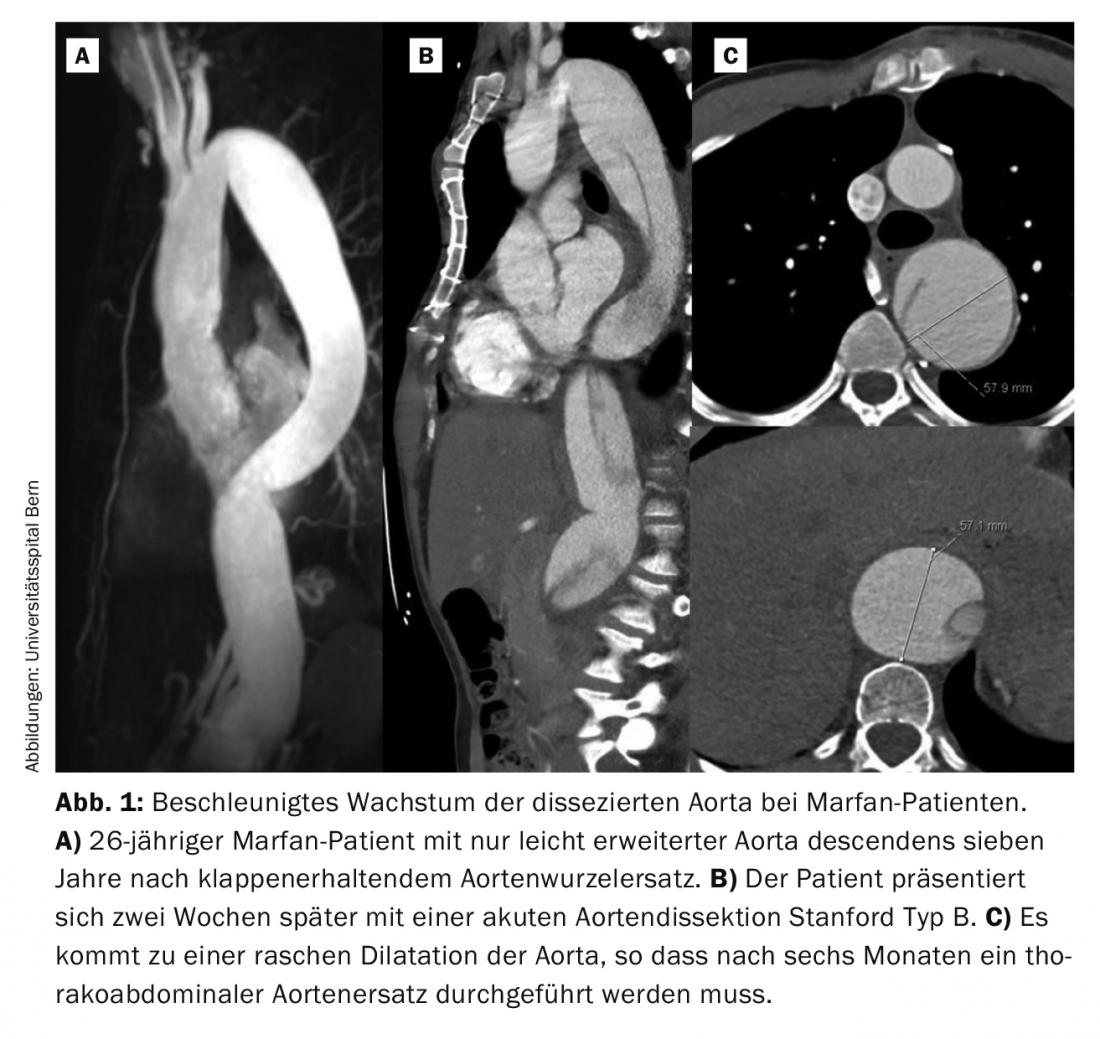
Tuttavia, spesso la manifestazione iniziale è la dissezione aortica acuta Stanford di tipo A o B. Nella nostra esperienza degli ultimi vent’anni, un terzo dei pazienti ha presentato una dissezione acuta. La dissezione acuta di tipo A è un’emergenza chirurgica e deve essere trattata immediatamente. Anche con un intervento riuscito, la metà di questi pazienti deve essere rioperata, per lo più sull’aorta distale, cioè non sostituita. Nei pazienti con sostituzione elettiva della radice aortica, solo il 10% circa dei pazienti deve ripetere l’intervento. Nella maggior parte dei casi, si tratta di pazienti che hanno subito una dissezione aortica di tipo B nel frattempo. Sebbene la dissezione aortica di tipo B sia spesso inizialmente “non complicata”, cioè senza malperfusione, nella stragrande maggioranza dei pazienti Marfan l’aorta deve essere successivamente sostituita toracoabdominalmente. Tipico nei pazienti Marfan è una rapida crescita dell’aorta dissecata nelle prime settimane e nei primi mesi (Fig. 1) . L’esperienza dimostra che il 50% dei pazienti richiede un intervento chirurgico nel primo anno dopo l’evento [6].
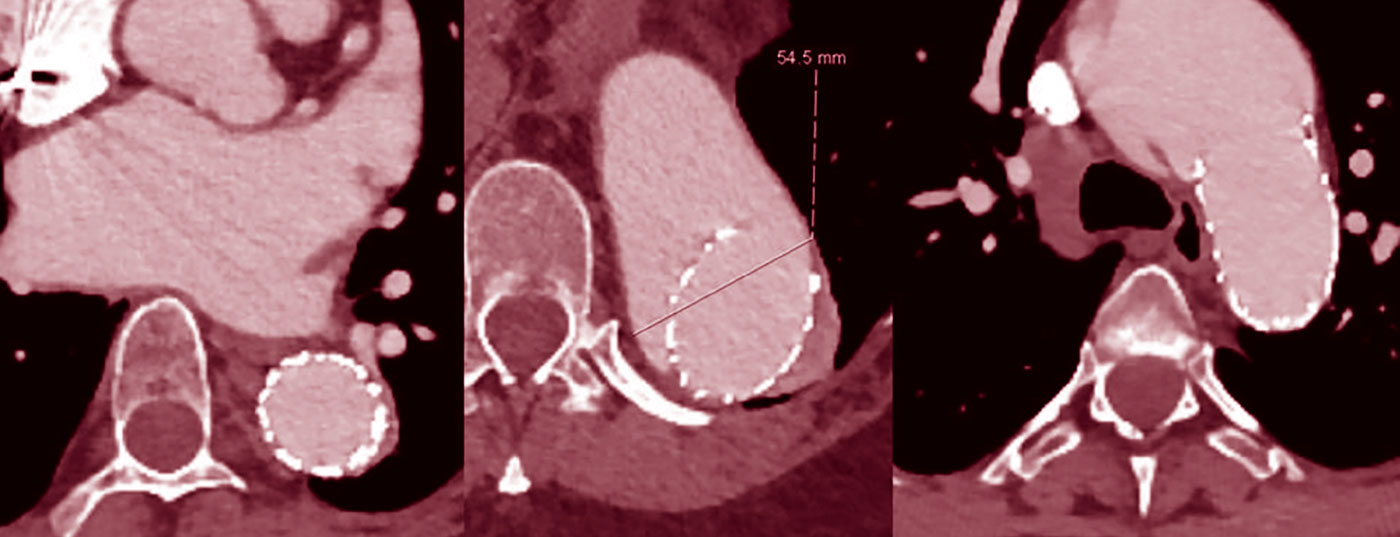
Il trattamento dei segmenti aortici toracoaddominali dilatati con innesti di stent è generalmente sconsigliato perché i segmenti dei vasi della zona di atterraggio spesso si dilatano secondariamente (Fig. 2), anche se inizialmente c’è un buon rimodellamento [7].

Opzioni farmacologiche
La dilatazione e il conseguente rischio di dissezione dell’aorta è la principale causa di morbilità e mortalità nei pazienti con malattie ereditarie del tessuto connettivo. L’obiettivo del trattamento farmacologico è quello di ridurre la progressione della dilatazione e il verificarsi di dissezioni. La riduzione della pressione arteriosa ai valori sistolici massimi di 120 mmHg e il tasso di aumento della pressione nella regione aortica ad ogni battito cardiaco sono di importanza rilevante. Tradizionalmente, il beta-bloccante è il farmaco di scelta [8]. Gli studi hanno dimostrato un tasso inferiore di dilatazione nei pazienti in terapia con beta-bloccanti, ma senza significato clinico in termini di sopravvivenza [9].
Negli ultimi anni, il bloccante del recettore ATII, il losartan, è stato studiato in modo più approfondito. Poiché interferisce direttamente con la via di segnalazione del TGFβ, si ripongono grandi speranze nel suo effetto per quanto riguarda la prevenzione della dilatazione e della dissezione aortica [10]. Tuttavia, un ampio studio randomizzato su bambini con sindrome di Marfan non ha rilevato alcuna differenza tra i beta-bloccanti e il losartan [11]. Lo studio è stato controverso perché in definitiva ha confrontato una dose molto alta di atenololo con una dose relativamente bassa di losartan. Grazie al profilo di effetti collaterali più basso, l’antagonista ATII è quindi la nostra prima scelta per il trattamento dei pazienti naïve alla terapia con una funzione di pompa normale. Gli antagonisti ATII sono ben tollerati, soprattutto da bambini e adolescenti. Studi più piccoli hanno dimostrato che la pressione sanguigna non si riduce in modo significativo in questa popolazione di pazienti [12].
Controlli di avanzamento
Per individuare tempestivamente l’insorgenza o la progressione di un aneurisma aortico ed evitare la dissezione aortica con una sostituzione aortica profilattica precoce, è essenziale effettuare un attento monitoraggio [13]. È importante valutare sistematicamente tutti i segmenti aortici. Inoltre, i controlli dopo l’intervento aortico sono importanti per individuare precocemente possibili complicazioni come la formazione di aneurismi in altre sezioni dell’aorta e le dissezioni asintomatiche.
Nelle nostre ore di consultazione, i pazienti vengono controllati post-operatoriamente dopo tre e dodici mesi mediante angio-TC e successivamente, a seconda dei risultati, una o tre volte all’anno. Per ridurre la dose cumulativa di radiazioni, questo dovrebbe essere fatto principalmente con la risonanza magnetica. L’esame di risonanza magnetica offre anche il vantaggio dell’imaging funzionale per quanto riguarda la funzione della pompa, la viziatura della valvola e la diagnostica dell’ischemia. Le operazioni precedenti con impianti sono raramente un problema quando si valutano le immagini. L’angiografia TC è attualmente ancora superiore alla risonanza magnetica nella valutazione delle dissezioni. I controlli ecocardiografici vengono eseguiti in caso di vitia o st.n.. Sostituzione della valvola eseguita una volta all’anno o alternativamente con esame di risonanza magnetica.
Una situazione particolare nell’assistenza a lungo termine dei pazienti con malattie ereditarie del tessuto connettivo è il desiderio di avere figli. Le prove sono poche e le raccomandazioni delle società professionali sono in parte contraddittorie [5,14,15]. In generale, un diametro aortico di <40 mm è considerato un rischio accettabile di gravidanza, sotto stretto controllo ecocardiografico e con il beta-blocco. Per i diametri >45 mm, è chiaramente consigliata la chirurgia profilattica. Per i diametri compresi tra 40 e 45 mm, la situazione individuale del paziente deve essere valutata con molta attenzione. Il fattore di rischio in questo caso è certamente un’anamnesi familiare positiva per quanto riguarda le dissezioni, soprattutto durante la gravidanza.
Le attuali linee guida raccomandano lo screening di tutti i parenti di primo grado in un paziente con aneurisma dell’aorta toracica. Questo passo può contribuire in modo significativo a ridurre il tasso di pazienti con dissezioni.
Messaggi da portare a casa
- Nei pazienti con malattie ereditarie del tessuto connettivo, l’indicazione per la sostituzione aortica profilattica viene data già con diametri di 45-50 mm.
- La valutazione regolare e sistematica di tutti i segmenti aortici previene le dissezioni e le rotture.
- Tutti i parenti di primo grado dei pazienti con aneurisma dell’aorta toracica devono essere sottoposti a screening per verificarne la presenza.
Letteratura:
- Brownstein AJ, et al: Geni associati all’aneurisma e alla dissezione dell’aorta toracica: aggiornamento 2018 e implicazioni cliniche. Aorta (Stamford) 2018; 6: 13-20.
- Habashi JP, et al: Il Losartan, un antagonista AT1, previene l’aneurisma aortico in un modello murino della sindrome di Marfan. Science 2006; 312: 117-121.
- Loeys BL, et al: Sindromi aneurismatiche causate da mutazioni nel recettore TGF-beta. N Engl J Med 2006; 355: 788-798.
- Pepin M, et al: Caratteristiche cliniche e genetiche della sindrome di Ehlers-Danlos di tipo IV, il tipo vascolare. N Engl J Med 2000; 342: 673-680.
- Erbel R, et al.: Linee guida ESC 2014 sulla diagnosi e il trattamento delle malattie aortiche: documento che copre le malattie aortiche acute e croniche dell’aorta toracica e addominale dell’adulto. La Task Force per la diagnosi e il trattamento delle malattie dell’aorta della Società Europea di Cardiologia (ESC). Eur Heart J 2014; 35: 2873-2926.
- Schoenhoff FS, et al: La dissezione aortica acuta determina il destino dei segmenti aortici inizialmente non trattati nella sindrome di Marfan. Circolazione 2013; 127: 1569-1575.
- Grabenwöger M, et al: Riparazione Aortica Endovascolare Toracica (TEVAR) per il trattamento delle malattie aortiche: una dichiarazione di posizione dell’Associazione Europea di Chirurgia Cardio-Toracica (EACTS) e della Società Europea di Cardiologia (ESC), in collaborazione con l’Associazione Europea di Interventi Percutanei Cardiovascolari (EAPCI). Eur Heart J 2012; 33: 1558-1563.
- Shores J, et al: Progressione della dilatazione aortica e beneficio del blocco beta-adrenergico a lungo termine nella sindrome di Marfan. N Engl J Med 1994; 330: 1335-1341.
- Gersony DR, et al: L’effetto della terapia beta-bloccante sull’esito clinico nei pazienti con sindrome di Marfan: una meta-analisi. Int J Cardiol 2007; 114: 303-308.
- Habashi JP, et al.: La segnalazione del recettore dell’angiotensina II di tipo 2 attenua l’aneurisma aortico nei topi attraverso l’antagonismo di ERK. Science 2011; 332: 361-365.
- Lacro RV, et al: Atenololo rispetto a losartan nei bambini e nei giovani adulti con sindrome di Marfan. N Engl J Med 2014; 371: 2061-2071.
- Brooke BS, et al: Blocco dell’angiotensina II e dilatazione della radice aortica nella sindrome di Marfan. N Engl J Med 2008; 358: 2787-2795.
- Jondeau G, et al: Tasso di eventi aortici nella popolazione di Marfan, uno studio di coorte. Circolazione 2012; 125: 226-232.
- Baumgartner H, et al: Linee guida ESC per la gestione delle cardiopatie congenite dell’adulto (nuova versione 2010). Eur Heart J 2010; 31: 2915-2957.
- Hiratzka LF, et al.Linee guida ACCF/AHA/AATS/ACR/ASA/SCA/SCAI/SIR/STS/SVM 2010 per la diagnosi e la gestione dei pazienti con malattia aortica toracica: un rapporto dell’American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines, dell’American Association for Thoracic Surgery, dell’American College of Radiology, dell’American Stroke Association, della Società degli Anestesisti Cardiovascolari, della Società per l’Angiografia Cardiovascolare e gli Interventi, della Società di Radiologia Interventistica, della Società dei Chirurghi Toracici e della Società di Medicina Vascolare. Circolazione 2010; 121: e266-369.
CARDIOVASC 2018; 17(5): 27-29









