Essendo il disturbo della coagulazione ereditario più comune, la sindrome di Von Willebrand colpisce circa una persona su mille. Con le nuove possibilità diagnostiche e le scoperte nel campo della genetica molecolare, viene sempre più spesso sollevata la questione dell’importanza clinica dell’analisi genetica. Questo è stato anche un argomento molto dibattuto al 63° Meeting annuale della Società Americana di Ematologia (ASH).
Test genetici nella sindrome di von Willebrand (Malattia di von Willebrand, La vWD) è come l’amore, tutti ne parlano, ma nessuno lo capisce esattamente – con queste parole Emmanuel Favaloro, un rinomato ricercatore australiano della Istituti di patologia clinica e ricerca medica a Ospedale Westmead a Sydney, la sua conferenza su Riunione annuale ASH 2021. E in effetti c’è un po’ di incertezza sul modo migliore di procedere. Mentre l’analisi genetica non ha senso nella maggior parte dei casi di sindrome di von Willebrand di tipo 1, è raccomandata per i pazienti di tipo 2 e 3 nelle nuove linee guida pubblicate nel 2021 [2].
Sindrome di von Willebrand: le basi
La sindrome di Von Willebrand è stata descritta per la prima volta nel 1926 come “pseudoemofilia” in una giovane donna che aveva sanguinato a morte durante una delle prime mestruazioni. Lo scopritore – nomen est omen – è il dottor Erik von Willebrand. La malattia ereditaria autosomica è definita da anomalie quantitative o qualitative del fattore von Willebrand (vWF); la prevalenza sintomatica è di circa 1/1000. Sebbene uomini e donne siano formalmente colpiti allo stesso modo, le donne ricevono la diagnosi da due a tre volte più spesso a causa delle complicazioni ginecologiche. Nella maggior parte dei casi, la sindrome di von Willebrand si manifesta con un aumento del sanguinamento mucoso; nei casi più gravi, può verificarsi anche un sanguinamento muscolo-scheletrico.
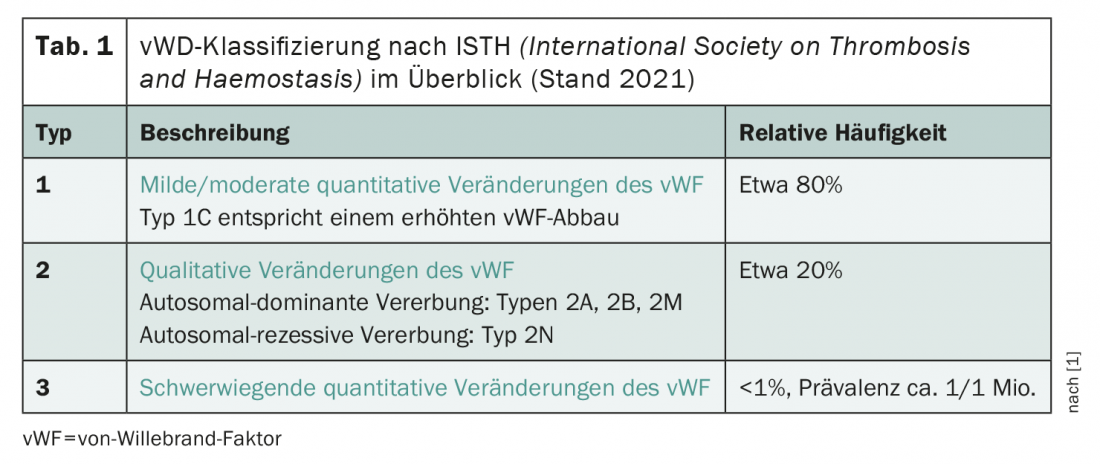
Secondo la classificazione recentemente rivista della Società Internazionale sulla Trombosi e l’Emostasi (ISTH), il quadro clinico è approssimativamente diviso in tre tipi (tab. 1) . In linea di principio, si distingue tra qualitativo (tipo 2) e cambiamenti quantitativi (tipi 1 e 3) del vWF. Nel 2021, è stato aggiunto alla classificazione il sottotipo 1C, definito da una maggiore degradazione del vWF. Il tipo 1 La sindrome di von Willebrand – cioè le alterazioni quantitative da lievi a moderate del vWF – è di gran lunga la più frequentemente diagnosticata, seguita dal tipo 2. L’assenza quasi completa di vWF nella sindrome di von Willebrand di tipo 3 è estremamente rara e colpisce circa una persona su un milione.
Oltre alla terapia sintomatica con farmaci antifibrinolitici come il Cyklokapron e la pillola nelle pazienti donne, esistono anche opzioni di trattamento che mirano direttamente al vWF. Si tratta di desmopressina (DDAVP), che promuove il rilascio di vWF dalle cellule endoteliali, e di vWF ricombinante o derivato dal plasma sanguigno. In linea di massima, la terapia per tutti i tipi di sindrome di von Willebrand è simile e dipende dalla gravità. Nei casi più gravi, il vWF di solito deve essere sostituito, poiché l’effetto del DDAVP diminuisce rapidamente già dopo un breve periodo di trattamento.
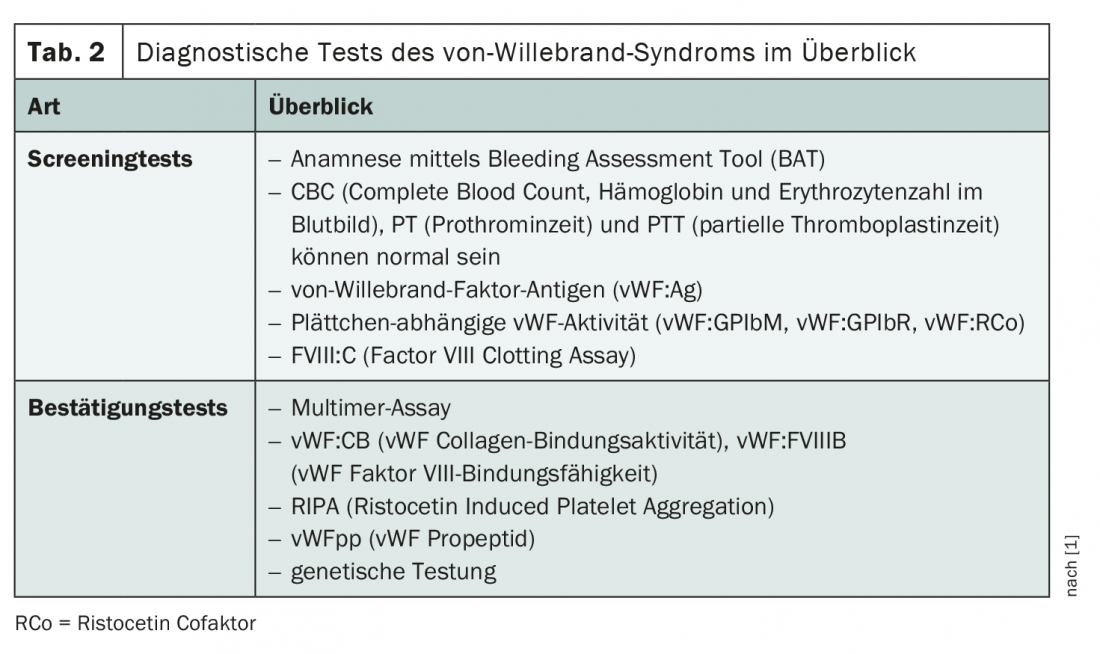
Per la diagnosi si utilizzano diversi test di screening e di conferma (tab. 2). Anche l’anamnesi medica gioca un ruolo decisivo. Poiché l’emocromo, il tempo di protrombina e il tempo di tromboplastina parziale possono essere normali, sono necessarie procedure di screening più specifiche. Di solito viene eseguito un pannello di tre strumenti diagnostici per rilevare gli antigeni vWF, l’attività vWF piastrino-dipendente e l’attività del fattore VIII. Se il sospetto di sindrome di von Willebrand viene confermato durante queste indagini, si ricorre a ulteriori test per confermare e identificare il sottotipo di vWD.
I test genetici sono controversi
Nel contesto di ulteriori test, anche l’analisi genetica svolge un ruolo finora controverso. Poiché la terapia non varia tra i sottotipi e sono disponibili metodi alternativi e più favorevoli per classificare la malattia, si pone la questione dell’utilità della caratterizzazione genetica. La loro disponibilità varia, ma è in aumento. Non bisogna trascurare eventuali effetti negativi sulle persone interessate, perché i test genetici possono comportare problemi assicurativi e stigmatizzazione, ad esempio. La conclusione è che l’analisi genetica del sottotipo più comune di vWD, il tipo 1, di solito non è utile, secondo gli esperti presenti alla riunione annuale dell’ASH – tra l’altro, perché una variante genetica corrispondente può essere rilevata solo in circa il 65% dei casi e questo non ha alcun impatto sulla gestione.
Anche se è stato fatto molto in termini di caratterizzazione genetica della malattia, che è stata raggiunta per la prima volta nel 1984, il database è ancora pieno di errori e lacune. Per esempio, esistono alcune cosiddette “varianti di incerto significato clinico” e alcune alterazioni genetiche non possono ancora essere chiaramente assegnate ai loro fenotipi corrispondenti. Tuttavia, un vantaggio dei test genetici è la loro fattibilità centralizzata e quindi le buone possibilità di standardizzazione. Il gene vWF è localizzato sul braccio corto del cromosoma 12.
Quando è utile l’analisi genetica?
Nonostante tutte le riserve, ci sono alcune situazioni in cui il test genetico nella sindrome di von Willebrand è già oggi molto apprezzato. Soprattutto nel caso dei sottotipi 2B, 2N e 3 di vWD, può fornire informazioni aggiuntive importanti e clinicamente rilevanti – da un lato per la differenziazione da altri disturbi della coagulazione e dall’altro per la consulenza genetica e la valutazione del rischio terapeutico. Le mutazioni possono essere rilevate in circa il 90% dei casi di sindrome di von Willebrand di tipo 2, e in circa l’85% dei casi di tipo 3. Per quanto riguarda la procedura diagnostica ottimale, comprese le analisi genetiche, nel 2021 sono state pubblicate nuove linee guida [2].
Questi raccomandano il test genetico per i sospetti sottotipi 2A, 2B e 2N di vWD [2]. La questione principale qui è la demarcazione dal cosiddetto “Le piastrine sindrome di von Willebrand” (nota anche come “pseudotipo”) e l’emofilia A. Questo perché la sindrome di von Willebrand di tipo 2B – tipicamente caratterizzata da un’aumentata affinità per le piastrine, da una cospicua analisi multimer e da trombocitopenia – assomiglia fortemente alla sindrome di von Willebrand di tipo 2B. Di tipo piastrinico. Tuttavia, mentre in quest’ultima la mutazione scatenante è localizzata nel gene delle piastrine, nella sindrome di von Willebrand di tipo 2B è localizzata nel gene vWF. Questo ha importanti implicazioni per la gestione: la “vera” sindrome di von Willebrand viene trattata con la sostituzione del vWF, lo pseudotipo con la somministrazione di piastrine. Il tipo piastrinico è ereditato in modo autosomico dominante. La causa è una mutazione con guadagno di funzionedella glicoproteina che lega il vWF. Questo porta a un’interazione eccessiva e non necessaria tra le piastrine e il vWF, con un consumo consecutivo di entrambi i componenti – e un quadro clinico che spesso assomiglia al più comune tipo di sindrome di von Willebrand “vera”. Il 2B è confuso. Si stima che circa il 15% del tipo 2B diagnostica in verità una sindrome di von Willebrand di tipo piastrinico [3].
Esiste anche una diagnosi differenziale importante per la sindrome di von Willebrand di tipo 2N (“Normandia”): l’emofilia A. In questo sottotipo, i livelli di fattore VIII sono classicamente più bassi dei livelli di vWF – una costellazione che si riscontra solo nella sindrome di von Willebrand nel caso del tipo Si verifica la 2N ed è associata all’emofilia. A può essere confuso. Pertanto, le nuove linee guida raccomandano che, nei casi di sospetto di tipo 2N vWD il test genetico mirato e/o la determinazione della capacità di legame del fattore VIII vWF [2]. Solo identificando correttamente la malattia di base, si può garantire una terapia adeguata, attraverso la sostituzione del fattore VIII o del vWF.
A differenza della sindrome di von Willebrand di tipo 2, la diagnosi del tipo 3 di solito vengono chiariti prima del test genetico. Tuttavia, può avere senso in questa rara forma autosomica recessiva della malattia – anche se la localizzazione della mutazione scatenante varia maggiormente rispetto ai tipi geneticamente più chiaramente caratterizzati. 2A, B e N. Da un lato, l’analisi genetica può svolgere un ruolo importante nella consulenza genetica delle persone affette e dei loro familiari, dall’altro fornisce informazioni preziose per la valutazione del rischio di una terapia con vWF esogeno. Quindi, le grandi delezioni predispongono alla formazione di allo-anticorpi durante la terapia. I pazienti con mutazioni nel propeptide vWF sembrano avere anche un rischio maggiore di emorragia rispetto a quelli con mutazioni in altre sedi.
La conclusione è che il beneficio del test genetico nel tipo 1 sindrome di von Willebrand – e quindi in oltre il 70% di tutte le persone colpite – è attualmente discutibile. Oltre alla correlazione genotipo-fenotipo non sufficientemente caratterizzata, manca anche la conseguenza terapeutica di tali analisi. Per i tipi 2 e 3, invece, l’analisi genetica può essere molto utile ed è già molto apprezzata nella diagnostica differenziale, nella valutazione del rischio e nella consulenza genetica.
Congresso: 63a Conferenza annuale ASH
Fonte/letteratura:
- Sessione Spotlight “Test genetici per la malattia di von Willebrand”, Paula James e Emmanuel Favaloro, 13.12.2021, 63° Meeting annuale ASH, Atlanta, USA.
- James PD, et al: Linee guida ASH ISTH NHF WFH 2021 sulla diagnosi della malattia di von Willebrand. Blood Adv. 2021; 5(1): 280-300.
- Othman M: La malattia di von Willebrand di tipo piastrinico: un disturbo emorragico raro, spesso mal diagnosticato e sottodiagnosticato. Semin Thromb Hemost. 2011; 37(5): 464-469.
InFo ONCOLOGIA & EMATOLOGIA 2022; 10(1): 38-40