Le dermatosi bollose autoimmuni sono un gruppo clinicamente e immunopatologicamente eterogeneo di malattie autoimmuni rare. Nei pazienti anziani con prurito e vesciche sporgenti, il pemfigoide bolloso è in cima alla lista delle diagnosi differenziali. Nei pazienti con erosioni nell’area della mucosa orale (“stomatite aftosa resistente alla terapia”), eventualmente in combinazione con vesciche/erosioni sulla pelle cheratinizzata, la presenza di una dermatosi autoimmune bollosa deve essere sempre considerata come diagnosi differenziale. L’anamnesi di epistassi, disfagia e dispnea è essenziale in questo caso, così come un’ispezione dettagliata della mucosa congiuntivale e genitale. Il trattamento delle dermatosi bollose autoimmuni deve essere effettuato in centri specializzati. Con le nuove opzioni terapeutiche, come il rituximab e l’immunoadsorbimento, anche le forme gravi della malattia possono essere trattate bene nella maggior parte dei casi. La determinazione dell’antigene bersaglio non solo è importante per una diagnosi esatta, ma può anche indicare la presenza di un processo paraneoplastico.
Le dermatosi autoimmuni bollose sono un gruppo di malattie autoimmunologiche che hanno in comune la presenza di autoanticorpi contro molecole strutturali e di adesione della pelle o della mucosa. Come risultato della disregolazione dell’immunità umorale e cellulare adattativa, si sviluppano delle fessure che sono clinicamente associate a vesciche ed erosioni sulla pelle e/o sulle membrane mucose.
Per quanto riguarda la localizzazione della formazione della fessura, è possibile dividere le dermatosi bollose autoimmuni in due gruppi principali per orientarsi:
1. Dermatosi bollose autoimmuni con perdita di aderenza intraepiteliale
– Malattie del pemfigo
2. dermatosi bollose autoimmuni con perdita di aderenza subepiteliale
– Malattie da pemfigoide
– Dermatosi lineare IgA
– Epidermolisi bollosa acquisita
– Dermatite erpetiforme Duhring
Malattie del pemfigo
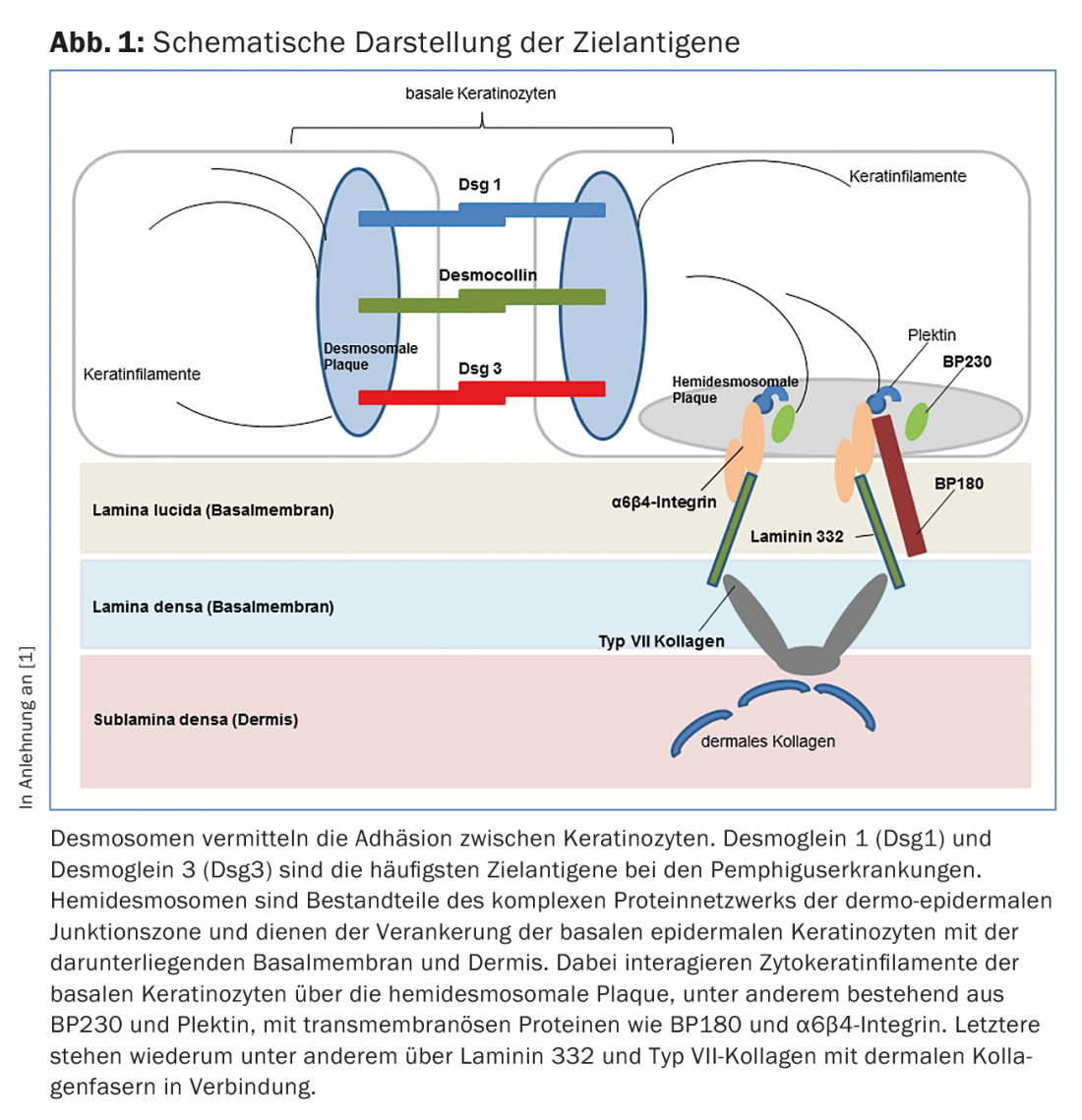
Nelle malattie del gruppo del pemfigo, la perdita del contatto intraepiteliale cellula-cellula si verifica a causa di autoanticorpi contro le proteine dei desmosomi (Fig. 1), con conseguente formazione di una fessura intraepidermica situata superficialmente. Dal punto di vista clinico e immunopatologico, si possono distinguere diverse forme di pemfigo.
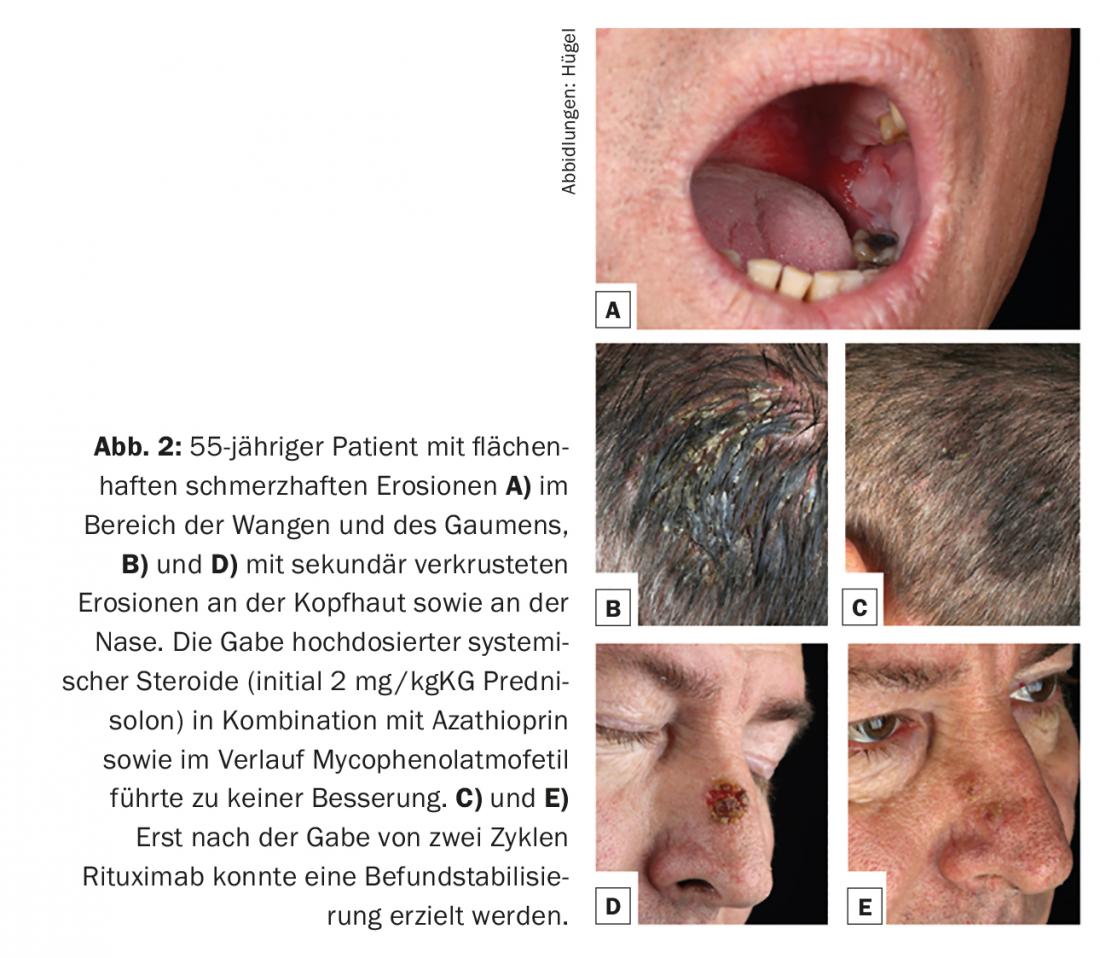
Il pemfigo vulgaris rappresenta circa l’80% dei casi di pemfigo e si manifesta preferibilmente tra la quarta e la sesta decade di vita, con un’incidenza di 0,1-0,5/100.000/anno. Dal punto di vista eziopatogenetico, si riscontrano sempre autoanticorpi contro la desmogleina 3. All’inizio della malattia, di solito sono presenti erosioni mucose enorali molto dolorose (Fig. 2A) e non è raro che la prima presentazione avvenga a uno specialista in malattie dell’orecchio, del naso e della gola. Facoltativamente, gli anticorpi contro la desmogleina 1 si trovano anche nel pemfigo vulgaris, con infestazione consecutiva della pelle cheratinizzata. (Fig. 2B e D). L’aspetto clinico della pelle è caratterizzato da vesciche estremamente vulnerabili con un tetto sottile, che si rompono rapidamente e quindi di solito non si presentano più come vesciche, ma come erosioni (secondariamente incrostate). Il fenomeno Nikolski I (spingibilità degli strati epidermici superiori mediante trazione tangenziale sulla pelle intatta) può essere indotto nell’esame clinico.
Al contrario, nella seconda malattia più comune del pemfigo – il pemfigo foliaceo – si trovano autoanticorpi solo contro la desmogleina 1, ma non contro la desmogleina 3. In accordo con il modello di espressione delle desmogleine, questa malattia colpisce quindi esclusivamente le aree cutanee cornee e non le membrane mucose. Anche in questo caso, non si trovano quasi mai vesciche intatte, ma erosioni estese con incrostazioni a volte simili a foglie. I sintomi cutanei spesso iniziano sulla testa pelosa, sul viso o sull’area del solco sudoriparo anteriore e posteriore e poi si diffondono alla periferia.
Il pemfigo paraneoplastico, che si verifica molto raramente, è associato in particolare a tumori maligni ematologici (soprattutto linfomi non-Hodgkin a cellule B) e presenta autoanticorpi contro bersagli sia desmosomiali che non desmosomiali. Clinicamente, è caratterizzata da estese erosioni e ulcere dolorose delle membrane mucose (soprattutto bocca, labbra, esofago), coinvolgimento congiuntivale e lesioni cutanee polimorfe.
Il pemfigo IgA (caratterizzato da depositi intraepidermici di IgA nella biopsia cutanea per l’immunofluorescenza diretta, Fig. 3C) è la più rara delle varianti di pemfigo. Clinicamente, le pustole e le vescicole su una base eritematosa si trovano principalmente nelle aree intertriginose (Fig. 3A e B).
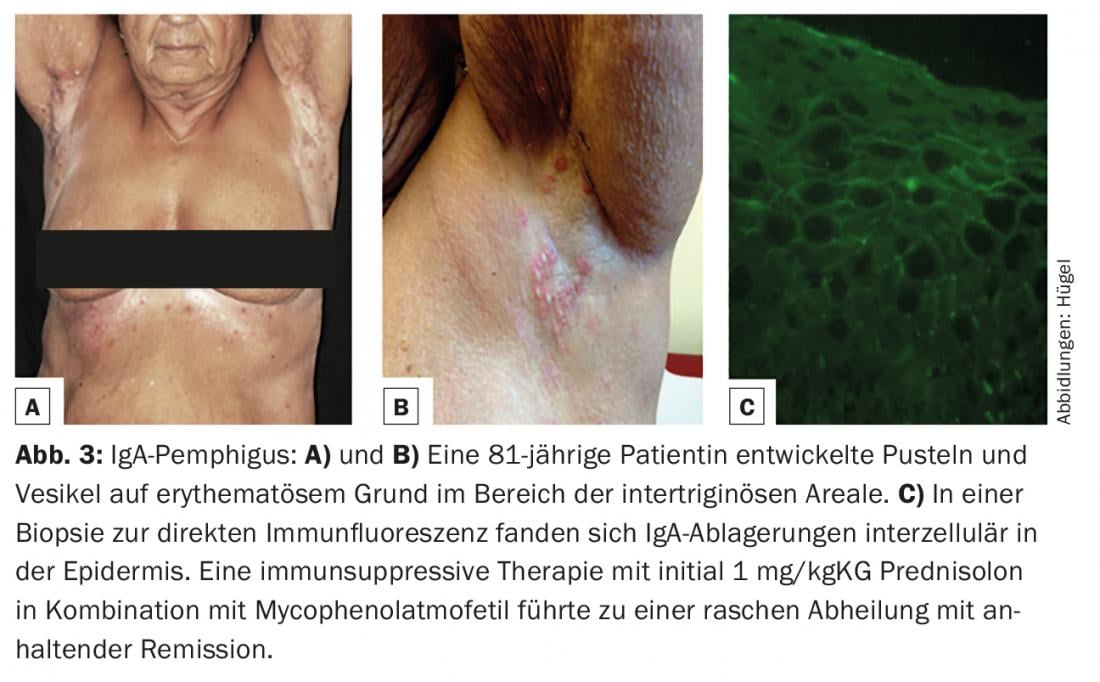
Tutti i pazienti con diagnosi di pemfigo devono avere un’attenta anamnesi farmacologica, in quanto farmaci come la penicillamina (che tuttavia oggi viene utilizzata molto raramente nel trattamento delle malattie reumatiche), ma anche gli ACE-inibitori come il captopril, l’enalapril o il lisinopril possono indurre la malattia del pemfigo.
Dermatosi autoimmuni vescicolose con perdita di adesione sottoepiteliale
Nelle malattie da pemfigoide, le vesciche sulla pelle appaiono molto più stabili e piene rispetto alle malattie da pemfigo, a causa della formazione di una fessura subepidermica più profonda. I pazienti spesso soffrono di un forte prurito. Il quadro clinico è eterogeneo.
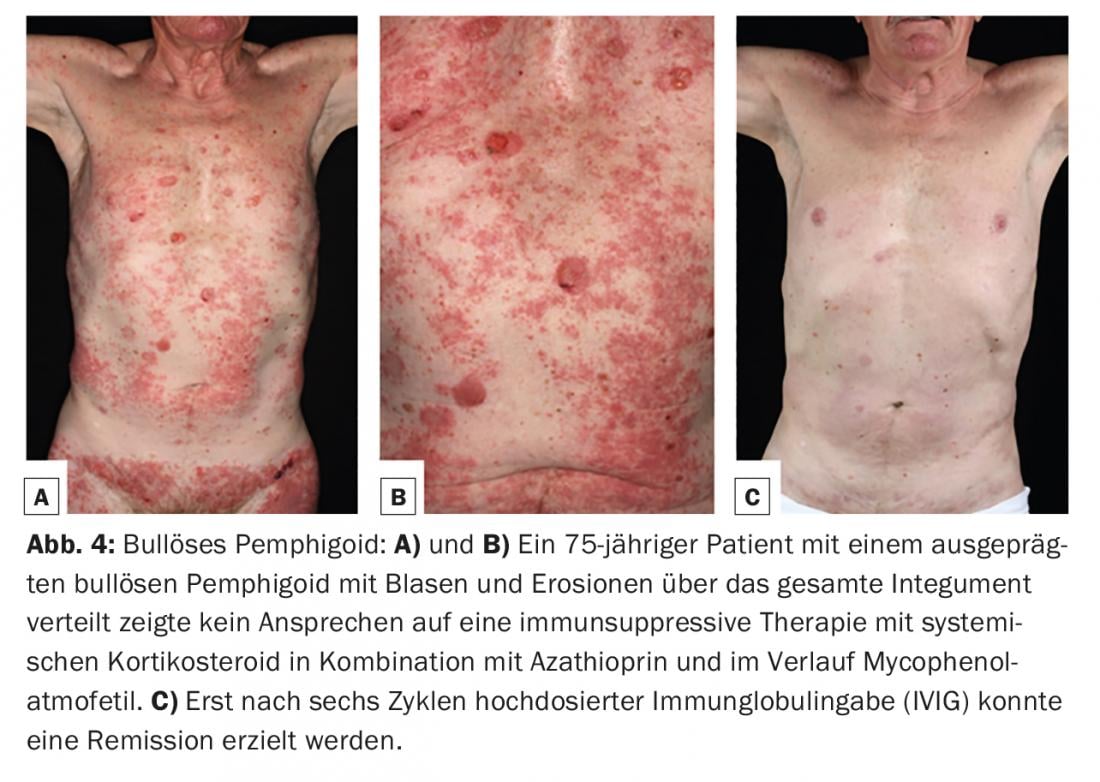
Il pemfigoide bolloso (BP) è la dermatosi autoimmune bollosa più comune, con un’incidenza di 12-21 casi per 1.000.000 di popolazione all’anno. L’età principale di insorgenza è tra i 60 e i 90 anni e, come risultato dell’aumento generale dell’aspettativa di vita, l’incidenza è aumentata notevolmente negli ultimi anni. Una caratteristica del pemfigoide bolloso è la presenza di autoanticorpi contro due proteine strutturali emidesmosomiali della zona della membrana basale: BP180 e/o (più raramente) BP230. (Fig.1). Clinicamente, il pemfigoide bolloso si presenta con vesciche rigonfie piene di liquido sieroso, eritema diffuso e lesioni orticarioidi seguite da erosioni e croste. (Fig. 4A e B). Le membrane mucose sono colpite dal 10-30%. In una fase premonitrice, la malattia può progredire per mesi o anni senza formazione di vesciche. Nei pazienti anziani con prurito grave e lesioni cutanee polimorfe (focolai di eczema, placche orticarioidi, escoriazioni come conseguenza del prurito), uno stadio pre-bolloso di BP deve quindi essere sempre considerato come diagnosi differenziale.
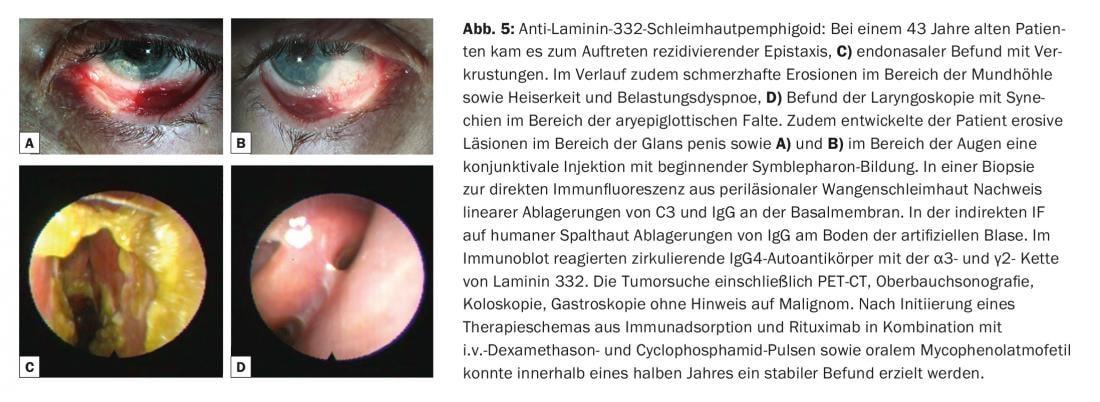
Il termine pemfigoide mucoso copre un gruppo eterogeneo di malattie rare dello spettro delle dermatosi autoimmuni bollose, che sono associate a vesciche subepiteliali o subepidermiche e in cui i cambiamenti infiammatori cronici si verificano prevalentemente nell’area delle membrane mucose. Le vesciche sono causate da autoanticorpi contro le molecole di adesione della zona di giunzione dermo-epidermica e sono stati identificati almeno dieci diversi antigeni bersaglio. Molto spesso, si riscontrano autoanticorpi IgG e/o IgA circolanti contro il BP180. Meno frequentemente, vengono rilevati autoanticorpi contro BP230, laminina-332, α6β4-integrina o collagene VII (Fig. 1). L’identificazione dell’antigene bersaglio è di grande importanza, in quanto è stata descritta un’associazione con i tumori maligni nel 30% dei casi per un sottotipo (pemfigoide anti-laminina-332) e deve essere eseguita una corrispondente esclusione del tumore. Clinicamente, tutte le membrane mucose con epitelio squamoso stratificato possono essere colpite nel pemfigoide mucoso. Il coinvolgimento più frequente è quello della mucosa orale, inizialmente con la comparsa di una gengivite desquamativa. Il coinvolgimento oculare di solito inizia unilateralmente con il quadro clinico della congiuntivite; nel corso del tempo, si verificano l’accorciamento delle pieghe congiuntivali (fornici), il symblepharon, la trichiasi, le sinechie e l’atrofia corneale, con il rischio finale di cecità. (Fig. 5A e B). Crosticine endonasali, epistassi, disfagia, raucedine e stridore si riscontrano con il coinvolgimento nasofaringeo o laringeo. (Fig. 5C e D). Negli uomini, le infestazioni genitali possono causare aderenze tra il glande del pene e il prepuzio e, nelle donne, l’ostruzione dell’introito vaginale. Il coinvolgimento cutaneo – clinicamente simile al pemfigoide bolloso – si riscontra in circa il 20% di tutti i casi.
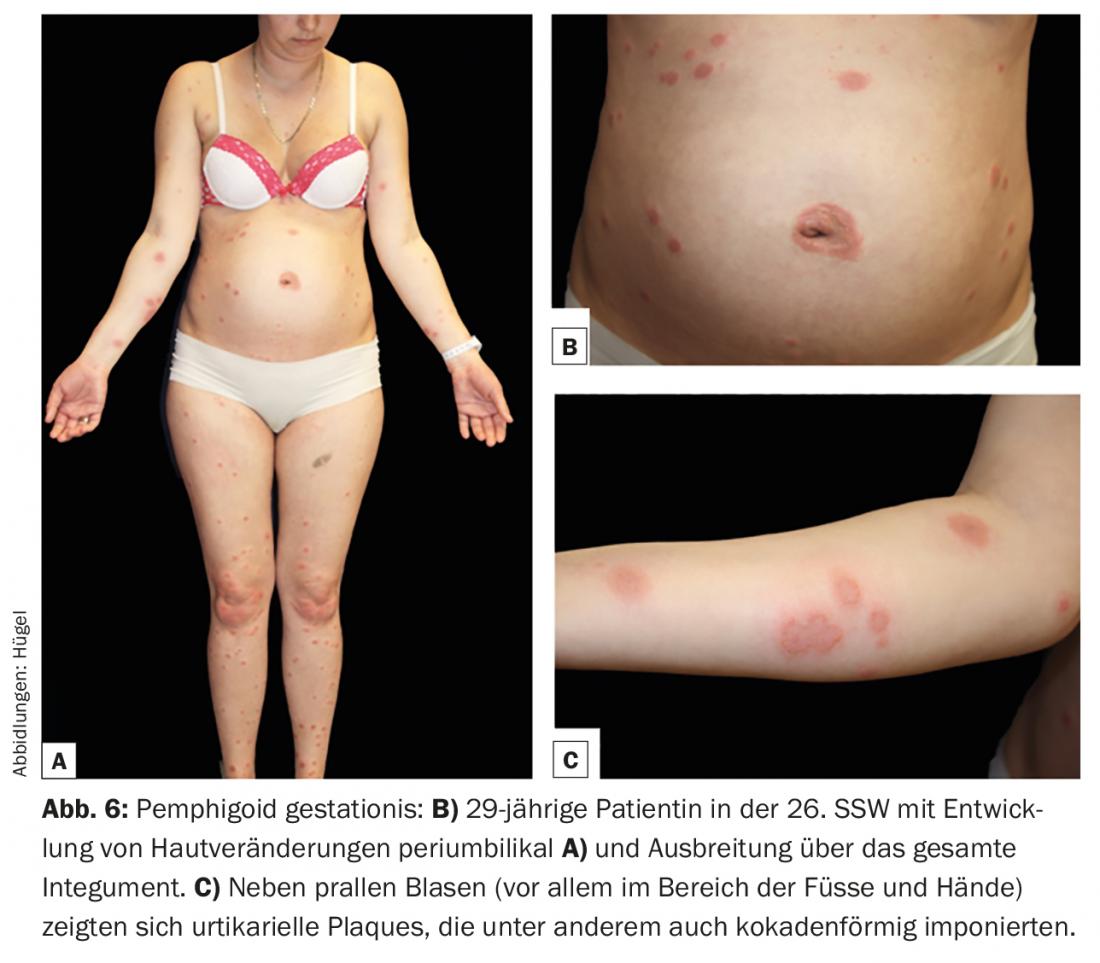
Il pemfigoide gestazionale (PG) è una rara dermatosi della gravidanza che può verificarsi nel secondo e terzo trimestre o subito dopo il parto ed è fisiopatologicamente molto vicina al pemfigoide bolloso. Come in questo caso, BP180 e – molto più raramente – BP230 sono gli antigeni target cruciali. La causa dello sviluppo degli autoanticorpi non è ancora stata chiarita. Il sistema HLA sembra svolgere un ruolo importante nella predisposizione genetica. Si discute sul fatto che la presentazione di autoantigeni placentari insieme alle molecole HLA paterne di classe II porta all’induzione di autoanticorpi. Clinicamente, lo sviluppo di efflorescenze specifiche è spesso preceduto da una fase prodromica con prurito intenso. Le lesioni cutanee di solito iniziano a livello periurbano (Fig. 6B) e possono poi diffondersi sull’intero tegumento. Le lesioni più comuni sono papule e/o placche orticarioidi, che talvolta appaiono a forma di coquette. Solo nel corso della malattia si sviluppano vescicole raggruppate o vescicole rigonfie nella maggior parte dei pazienti (Fig. 6).
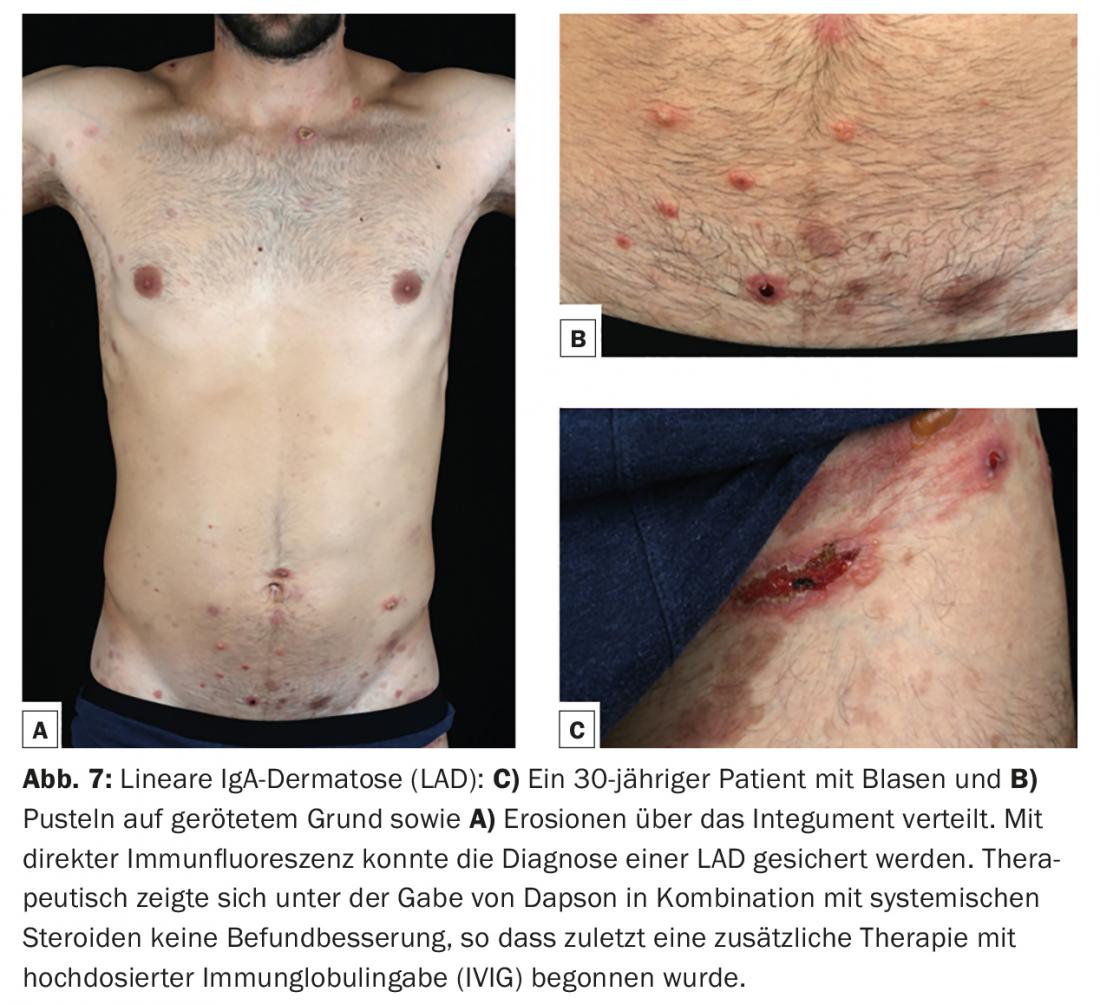
La dermatosi lineare IgA (LAD) è la dermatosi bollosa autoimmune più comune dell’infanzia e di solito inizia prima dei sei anni. In età adulta, la manifestazione iniziale è possibile a qualsiasi età, ma più spesso dopo i 60 anni. Come il pemfigoide mucoso, è considerato un quadro clinico eterogeneo. Sono caratteristici i depositi lineari di tipo IgA lungo la zona di giunzione dermo-epidermica nella biopsia cutanea per l’immunofluorescenza diretta. Gli autoanticorpi IgA sono più comunemente diretti contro una proteina di 97 kDa (LABD-97) e una proteina di 120 kDa (LAD-1), che sono prodotte dalla scissione proteolitica della parte extracellulare del BP180. Clinicamente, si presentano spesso vesciche anulari o policicliche sulla pelle sana o eritematosa, con un coinvolgimento preferenziale del viso (soprattutto periorale e delle orecchie), della regione anogenitale e, meno frequentemente, del tronco, delle mani e dei piedi. (Fig.7). Può verificarsi la cicatrizzazione delle membrane mucose, in particolare della congiuntiva, come nel pemfigoide mucoso, e può anche portare alla cecità.
Il quadro clinico dell’epidermolisi bollosa acquisita (EBA), in cui gli anticorpi di classe IgG (e IgA) contro il collagene VII sono eziopatogeneticamente presenti (Fig. 1) , varia notevolmente. Nella variante meccano-bollosa, la pelle è molto vulnerabile e si riscontrano vesciche ed erosioni che guariscono con cicatrici o milia, soprattutto nelle aree sottoposte a stress meccanico come il dorso della mano, il gomito, il ginocchio, la regione sacrale e le dita dei piedi. Nelle varianti infiammatorie, questa malattia presenta un quadro clinico che può essere difficile da distinguere dal pemfigoide bolloso o dal pemfigoide della mucosa.
La dermatite erpetiforme Duhring è un raro sottotipo cutaneo delle malattie sensibili al glutine. Qualche anno fa, la transglutaminasi epidermica (transglutaminasi 3) e la transglutaminasi tissutale (transglutaminasi 2) possono essere identificati. I pazienti affetti da questa malattia sviluppano papule e vescicole principalmente sui lati estensori delle estremità, sulla testa pelosa, a livello gluteo e sacrale. Si tratta di una condizione estremamente pruriginosa. Pertanto, la malattia di Duhring deve essere presa in considerazione in tutti i pazienti con escoriazioni da grattamento in primo piano nell’area dei siti di predilezione sopra citati.
Diagnosi delle dermatosi autoimmuni bollose
Le biopsie cutanee per l’esame istologico convenzionale possono essere utilizzate per mostrare la localizzazione della fessura.
Tuttavia, il gold standard diagnostico è il rilevamento degli autoanticorpi legati al tessuto (IgG, IgA e/o fattore C3 del complemento) nelle biopsie della pelle o della mucosa con l’immunofluorescenza diretta. La localizzazione degli anticorpi a livello intercellulare nell’epidermide o lungo la zona di giunzione dermo-epidermica consente una differenziazione immediata tra la presenza di una malattia da pemfigo o una malattia dello spettro delle dermatosi autoimmuni bollose subepiteliali. Se predominano i precipitati dell’isotipo IgA, si può diagnosticare un pemfigo IgA (fig. 3C), una dermatosi lineare IgA o una malattia di Duhring, a seconda del modello (tab. 1).
Oltre alle biopsie cutanee (di muco) e per l’esatta caratterizzazione dei diversi sottotipi, sono necessari esami sierologici. L’immunofluorescenza indiretta sull’esofago di scimmia e sulla pelle salina umana è stata stabilita come test di screening per questo scopo. Gli antigeni target vengono poi identificati con l’aiuto di vari ELISA e, se necessario, di esami specifici di immunoblot.
Le concentrazioni di autoanticorpi nel siero dei pazienti affetti da pemfigo e pemfigoide di solito sono ben correlate con l’attività della malattia e sono quindi adatte anche per monitorare l’attività della malattia e valutare la necessità di un’ulteriore terapia.
Terapia delle dermatosi autoimmuni bollose
In generale, la terapia delle dermatosi autoimmuni bollose dipende dalla gravità, da un lato, e dal sottotipo diagnosticato, dall’altro. Nel caso, ad esempio, di un lieve pemfigoide bolloso localizzato, può essere sufficiente un trattamento locale solo con steroidi di classe IV altamente potenti (clobetasolo propionato 0,05% unguento). Nella maggior parte dei casi, tuttavia, è indicata la somministrazione di corticosteroidi sistemici in combinazione con altri immunosoppressori (ad esempio, azatioprina, micofenolato mofetile, ciclosporina, metotrexato, ciclofosfamide). Il dapsone è l’agente di prima linea per la dermatosi lineare IgA, la dermatite erpetiforme di Duhring e il pemfigoide mucoso orale puro non complicato. Nei casi gravi o refrattari, si può ricorrere alle immunoglobuline per via endovenosa (IVIG), all’immunoadsorbimento e/o al rituximab. Inoltre, negli ultimi anni sono stati pubblicati diversi casi che dimostrano l’efficacia della terapia con l’anticorpo anti-IgE omalizumab nel pemfigoide bolloso. Tuttavia, questo richiede ulteriori studi di standardizzazione per quanto riguarda il dosaggio e la durata di utilizzo.
La cura dei pazienti con dermatosi autoimmuni bollose deve avvenire in centri specializzati e richiede – soprattutto in presenza di un sottotipo con coinvolgimento mucoso – un approccio interdisciplinare con il coinvolgimento di oculisti, otorinolaringoiatri, dentisti, ginecologi, gastroenterologi e lo specialista dermatologo come coordinatore.
Letteratura:
- Schmidt E, Zillikens E: Diagnostica e terapia delle dermatosi autoimmuni bollose. Dtsch Arztebl International 2011; 108(23): 399-405.
Ulteriori letture:
- Hertl M (ed.): Malattie autoimmuni della pelle. Patogenesi, diagnosi, gestione.Terza edizione. Vienna – New York: Springer-Verlag 2011.
- Marazza G, et al: Incidenza del pemfigoide bolloso e del pemfigo in Svizzera: uno studio prospettico di 2 anni. Br J Dermatol 2009; 161(4): 861-868.
- Kneisel A, Hertl M: Malattie cutanee bollose autoimmuni. Parte 1: Manifestazioni cliniche. J Dtsch Dermatol Ges 2011; 9(10): 844-856.
- Kneisel A, Hertl M: Malattie cutanee bollose autoimmuni. Parte 2: diagnosi e terapia. J Dtsch Dermatol Ges 2011; 9(11): 927-947.
- Kneisel A, Hertl M: Pemfigoide bolloso: diagnosi e terapia. Wien Med Wochenschr 2014; 164(17-18): 363-371.
- Schmidt E, Zillikens D: Malattie da pemfigoide. Lancet 2013; 381(9863): 320-332.
- Hertl M, et al.: Raccomandazioni per l’uso del rituximab (anticorpo anti-CD20) nel trattamento delle malattie cutanee bollose autoimmuni. J Dtsch Dermatol Ges 2008; 6(5): 366-373.
PRATICA DERMATOLOGICA 2017; 27(1): 18-25