La tachicardia ventricolare polimorfa catecolaminergica (CPVT) è una rara malattia dei canali ionici senza cardiopatia strutturale macroscopicamente rilevabile. Si verifica esclusivamente nell’infanzia e nell’adolescenza e ha un alto tasso di mortalità se non viene trattata. Pertanto, la terapia del cuore strutturalmente poco appariscente è di particolare importanza.
La tachicardia ventricolare polimorfa catecolaminergica (CPVT) è una rara malattia dei canali ionici senza cardiopatia strutturale macroscopicamente rilevabile, con una prevalenza di 1:5000-10.000. Descritta per la prima volta nel 1975 [1], è stata caratterizzata come entità separata solo nel 1995 [2]. Caratteristica è la comparsa di extrasistoli ventricolari polimorfe e di tachicardia ventricolare (VT), nonché di VT bidirezionali in condizioni di stress fisico o mentale, con un ECG a riposo non significativo. Sebbene esistano casi individuali di prime manifestazioni sintomatiche fino all’età di 40 anni, si tratta prevalentemente di una malattia dei bambini e degli adolescenti. A seconda del sottotipo genetico, la prima manifestazione si verifica tra i 2 e i 20 anni di età, con un cluster familiare nel 30% dei casi. Uomini e donne sono ugualmente colpiti, con una presentazione più precoce negli uomini [3]. Lo screening familiare dovrebbe essere sempre effettuato [4].
Fisiopatologia
Sebbene ci siano già stati grandi progressi nella caratterizzazione dei meccanismi patologici sottostanti dalla descrizione iniziale, questi non sono ancora del tutto compresi.
I difetti nel rilascio di calcio (Ca2+) diastolico dal reticolo sarcoplasmatico (SR) determinano un sovraccarico di Ca2+ nella cellula miocardica. Negli individui sani, l’afflusso di Ca2+ extracellulare attraverso i canali di tipo L, innescato dal potenziale d’azione, determina il rilascio di Ca2+ dal SR attraverso l’attivazione del recettore della rianodina (RYR2). Questo porta alla contrazione miocardica e alla fase di plateau del potenziale d’azione, con il successivo trasporto di Ca2+ nello spazio extracellulare e nel SR. Nella CPVT, tuttavia, un difetto autosomico dominante di RYR2, tra gli altri, porta a un rilascio diastolico spontaneo di Ca2+ dal SR. Questo porta a un’inversione del trasporto di Ca2+ nella cellula e alle postdepolarizzazioni tardive, che costituiscono la base della CPVT. Se anche la stimolazione β-adrenergica è innescata dallo stress, si sviluppano le caratteristiche extrasistoli o tachicardie ventricolari polimorfiche [5].
Oltre alle mutazioni nel gene RYR2, che sono rilevabili in circa il 50-55% dei pazienti con CPVT, esistono altre mutazioni della calsequestrina cardiaca (CASQ2) e della triadina (TRDN), due altri componenti del bilancio del Ca2+ cellulare [5]. Inoltre, anche la disfunzione dei canali del potassio (KCNJ2) può portare alla CPVT. A seconda del difetto genetico rilevato, si possono distinguere diversi sottotipi. In generale, tuttavia, i difetti genetici responsabili possono essere individuati solo in circa il 60% delle persone colpite [5].
Diagnosi
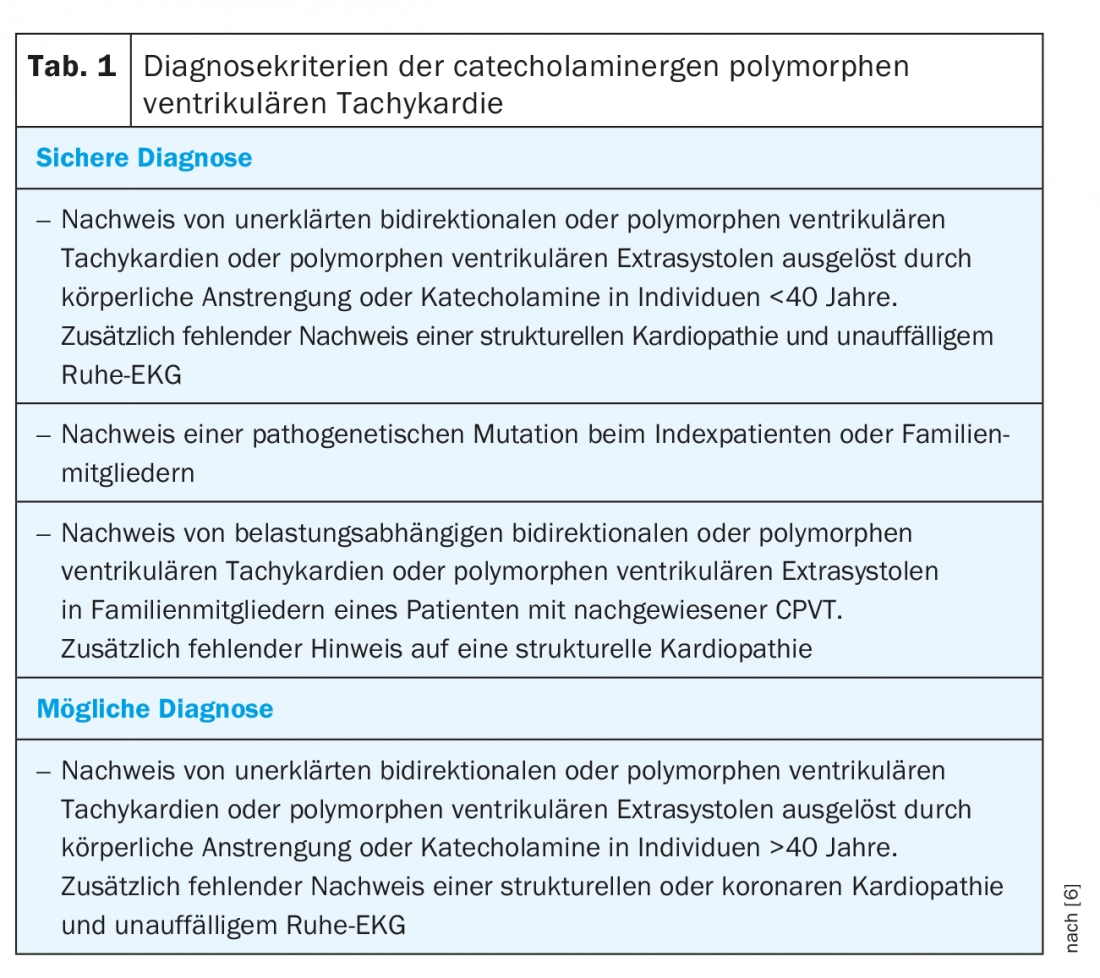
I criteri diagnostici delle società professionali sono riassunti nella tabella 1 [6]. Tuttavia, è molto più difficile che applicare i criteri diagnostici per identificare i possibili malati. Le presentazioni cliniche tipiche sono quelle di bambini di età compresa tra 2-20 anni che hanno avuto una sincope o sono sopravvissuti a una morte cardiaca improvvisa (SCD) durante uno stress fisico o emotivo [2]. Poiché la sincope legata alla CPVT può anche causare convulsioni e incontinenza, i bambini vengono spesso trattati erroneamente per l’epilessia. La CPVT viene quindi diagnosticata tardivamente, in assenza di efficacia dei farmaci antiepilettici. Allo stesso modo, un gruppo familiare di sincope da sforzo o SCD e di epilessie familiari refrattarie dovrebbe suggerire una possibile CPVT familiare. La presentazione atipica nel contesto dello screening cardiovascolare mediante ergometria è possibile in età adulta, ma molto meno frequente.
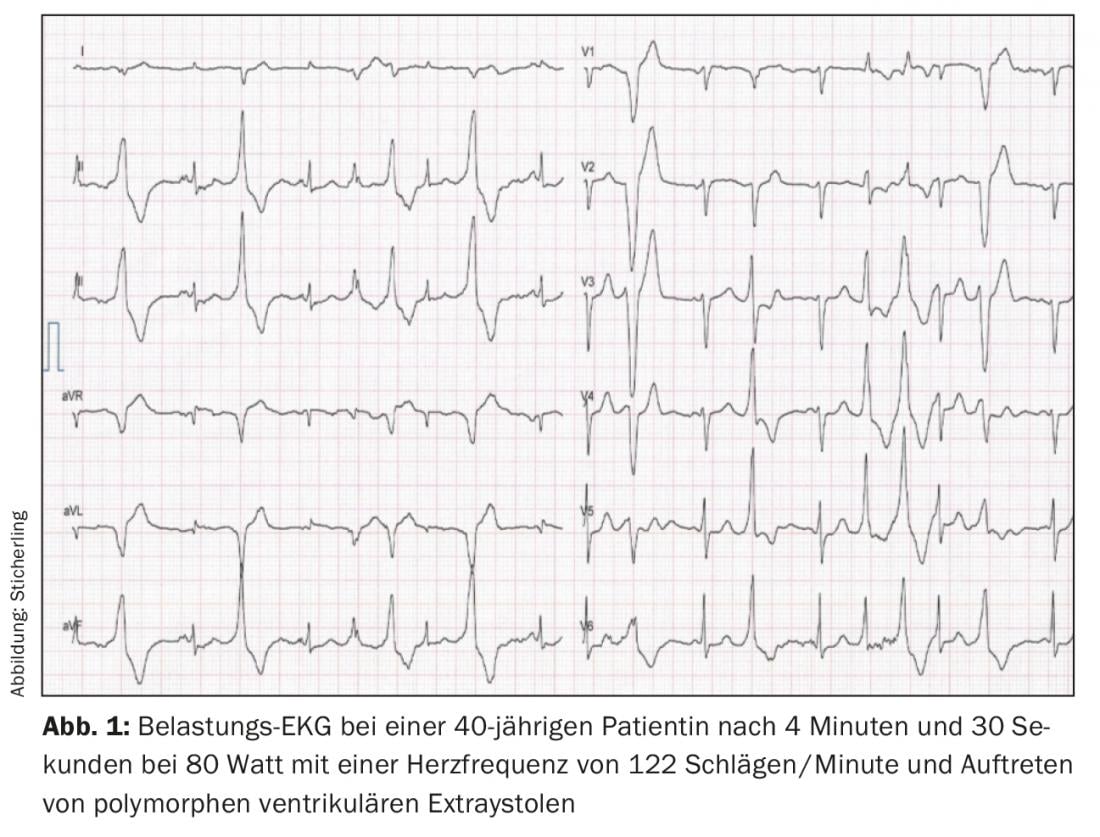
Il gold standard per la diagnosi è l’ergometria da sforzo. In genere, è possibile determinare una soglia di frequenza cardiaca riproducibile individualmente tra 110-130 battiti/minuto, al di sopra della quale si verificano le prime extrasistoli ventricolari isolate con un intervallo di accoppiamento di circa 400 ms. Le extrasistoli mostrano prevalentemente un asse superiore con blocco di branca sinistra o un asse inferiore con blocco di branca destra [4,7]. Con l’aumento dello stress e della frequenza cardiaca, si verificano extrasistoli monomorfe più frequenti, seguite da bigeminia e infine extrasistoli polimorfe e/o VT non sostenute (Fig. 1). Raramente, possono verificarsi VT sostenuti polimorfici o fibrillazione ventricolare. Il test da sforzo deve quindi essere interrotto se i sintomi aritmogeni aumentano o la durata dei VT non sostenuti aumenta [7,8]. Un ECG ambulatoriale a lungo termine può avere un valore diagnostico aggiuntivo nei pazienti molto giovani, nei pazienti che non possono eseguire un ECG da sforzo o nei pazienti con un ECG da sforzo negativo ma ancora sospettati di CPVT. Tuttavia, la sensibilità è inferiore rispetto all’ECG da sforzo [9]. Un’altra opzione diagnostica è l’infusione di catecolamine. Rispetto al test da sforzo, tuttavia, c’è una sensibilità inferiore, pari a circa il 75%, per cui dovrebbe essere utilizzato a scopo diagnostico solo in casi eccezionali [10]. L’esame elettrofisiologico con stimolazione programmata non ha alcun valore nella diagnosi di CPVT [2,10]. Come già detto, l’ECG a riposo e le tecniche di imaging non forniscono un valore diagnostico. Tuttavia, i risultati normali di questi esami sono obbligatori per poter fare la diagnosi [6].
La diagnosi differenziale deve includere altre malattie dei canali ionici come la sindrome del QT lungo, in particolare la SCD durante l’attività fisica come il nuoto [11]. Un ECG da sforzo può smascherare il prolungamento del QTc nella fase di recupero, che non è rilevabile nell’ECG a riposo, e quindi contribuire alla differenziazione dalla CPVT [12]. Un’altra diagnosi differenziale è la sindrome di Andersen-Tawil, che, come un sottotipo di CPVT, è associata a una mutazione in KCNJ2 e può anche presentarsi con tachicardia ventricolare bidirezionale [13]. Oltre ai segni fenotipici di paralisi periodica e dismorfia delle estremità, che non sono sempre presenti, anche l’ECG da sforzo può aiutare a differenziare questo caso dalla CPVT. La differenziazione è importante perché i pazienti con la sindrome di Andersen-Tawil hanno una prognosi più benigna [13]. Oltre alle malattie dei canali ionici, bisogna sempre pensare alle malattie strutturali non ancora diagnosticate, come le cardiomiopatie aritmogene, ipertrofiche, ischemiche o valvolari, che nel complesso sono molto più comuni. Altre cause di VT bidirezionale sono l’intossicazione da digossina o la miocardite [14,15].
Terapia
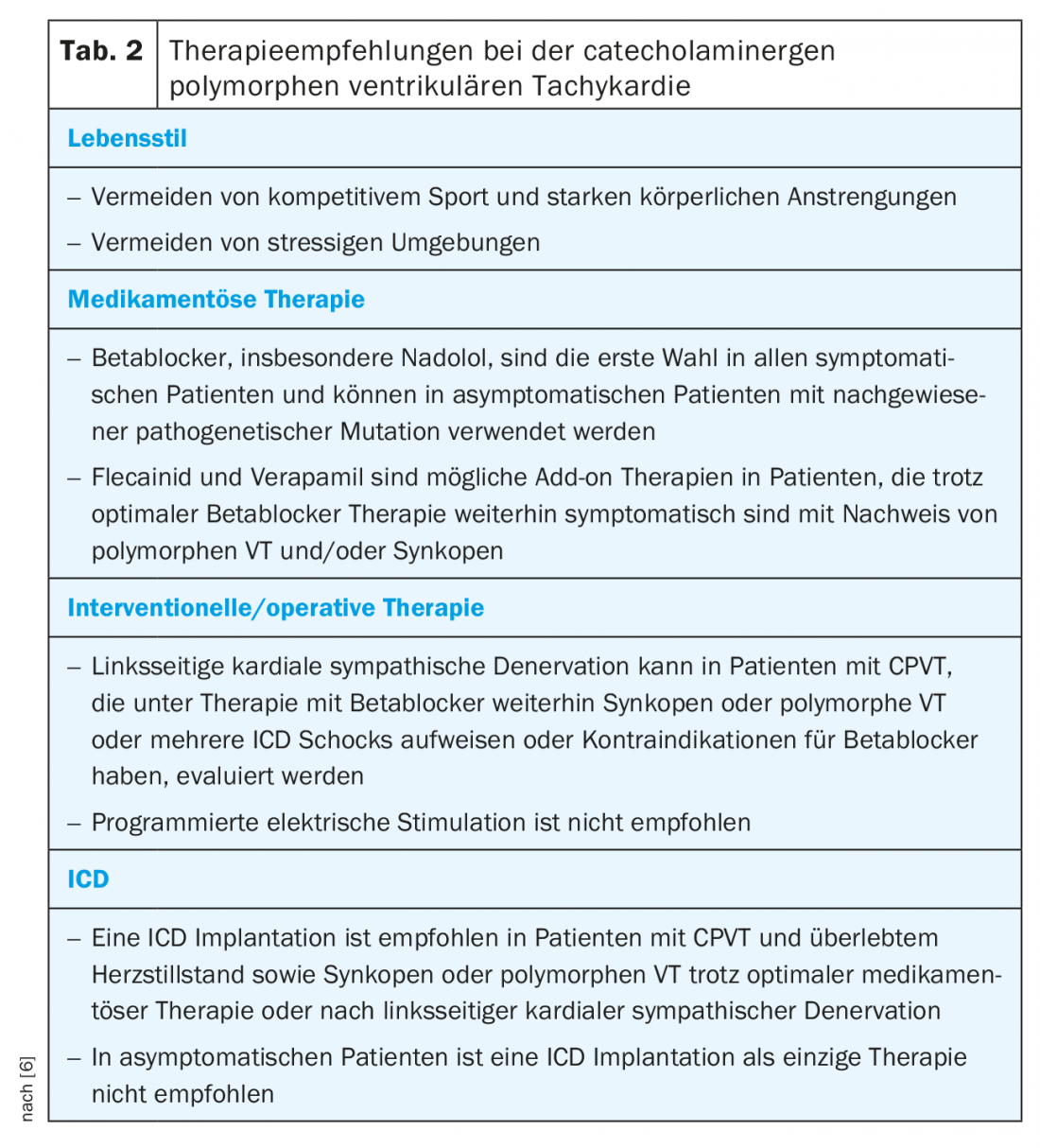
Il trattamento della CPVT consiste in cambiamenti dello stile di vita, terapia farmacologica, stratificazione del rischio per un ICD fino alla denervazione simpatica cardiaca sinistra (LKSD). Poiché la mortalità può raggiungere il 50% nei pazienti gravemente colpiti, una terapia adeguata è di grande importanza [2,16] (Tab. 2) . Inoltre, lo screening familiare deve essere sempre effettuato a causa dell’ereditarietà autosomica dominante del difetto RYR2.
Poiché lo sforzo fisico può scatenare aritmie ventricolari, i pazienti devono evitare l’esercizio agonistico. Un piccolo studio è stato in grado di dimostrare che nei pazienti con CPVT adeguatamente trattati, l’esercizio agonistico provoca solo un aumento delle aritmie senza un incremento della mortalità [17], ma la trasferibilità a tutti i pazienti non è possibile. Soprattutto quando si nuota, dovrebbe essere presente un supervisore.
I beta-bloccanti sono il pilastro della terapia farmacologica perché, tra le altre cose, impediscono il raggiungimento del limite aritmogeno della frequenza cardiaca. La maggior parte dei dati sono disponibili per il nadololo a lunga durata d’azione, non selettivo. Tuttavia, questa preparazione non è più disponibile in molti Paesi, come la Svizzera [16,18,19]. Un’alternativa è il carvedilolo, che ha un effetto comprovato su RYR2 [20]. Tuttavia, mancano dati clinici nei pazienti con CPVT. In linea di principio, si deve sempre somministrare un dosaggio completo fino alla dose massima tollerabile. È importante notare che nei bambini la dose deve essere adattata al peso corporeo. Una terapia beta-bloccante adeguata può ridurre il tasso di SCD fatale al 6,4% nell’arco di 8 anni [19]. Allo stesso modo, i pazienti positivi al genotipo, individuati dallo screening familiare, ma che non presentano aritmie all’ECG da sforzo, dovrebbero ricevere una terapia con beta-bloccanti [6]. Se le aritmie o gli accoppiamenti e le extrasistoli ventricolari a grappolo continuano ad essere osservati durante l’ergometria con una terapia beta-bloccante adeguata, si deve valutare una terapia aggiuntiva con flecainide. La flecainide mostra anche un effetto inibitorio diretto su RYR2 e negli studi clinici ha dimostrato di sopprimere le aritmie ventricolari nel 76% dei pazienti già trattati con beta-bloccanti [21,22]. I calcio-antagonisti sembrano avere un ulteriore beneficio come terapia aggiuntiva alternativa. Piccoli studi non randomizzati hanno dimostrato che il verapamil in aggiunta ai beta-bloccanti riduce ulteriormente il verificarsi di aritmie ventricolari [23,24]. Non sono ancora disponibili studi randomizzati più ampi sul verapamil. Le opzioni terapeutiche non ancora in uso clinico, ma potenzialmente promettenti, includono il propafenone, che si è dimostrato promettente in un numero limitato di pazienti [25] e il dantrolene, che ha dimostrato efficacia in un modello cellulare [26].
Se le aritmie non sono adeguatamente controllate con la terapia medica, si può valutare un LSD asportando toracoscopicamente la metà inferiore del ganglio stellato sinistro e i gangli toracici T2-4. Questo determina una marcata diminuzione della secrezione di noradrenalina nel cuore, con risultati molto buoni in piccole serie di pazienti con CPVT grave. La complicanza più frequente della LKSD è una sindrome di Horner per lo più transitoria, che tuttavia può persistere nel 2-3% dei casi. Complicazioni meno comuni sono le lesioni alle strutture circostanti, come la pleura, il nervo frenico, il plesso brachiale e i nervi somatici, con conseguente dolore lancinante alla spalla sinistra. Come effetto collaterale, può verificarsi una mancanza di sudorazione della mano sinistra e della fronte sinistra, con una pelle più calda e secca rispetto al lato [27,28].
L’impianto di ICD deve essere eseguito solo in pazienti selezionati che sono sopravvissuti a uno SCD o che continuano a manifestare sincope o VT polimorfica o bidirezionale nonostante la terapia ottimale. Se è disponibile un LKSD, anche questo dovrebbe essere eseguito prima dell’impianto [6]. Questa raccomandazione restrittiva esiste principalmente perché gli shock dell’ICD di per sé provocano un rilascio supplementare di catecolamine. Questi possono poi innescare altre aritmie e portare a un circolo vizioso, quindi l’ICD deve essere programmato con cut-off elevati e lunghi ritardi prima dell’erogazione dello shock [6,29].
Sommario
La CPVT è una malattia estremamente rara che si verifica quasi esclusivamente nei bambini e negli adolescenti e ha un alto tasso di mortalità se non viene trattata. La terapia con beta-bloccanti è l’intervento più importante in termini di prognosi, insieme all’adeguamento dello stile di vita. L’impianto di ICD è associato a effetti speciali, potenzialmente proaritmici con possibile erogazione di shock e l’indicazione nella profilassi primaria deve quindi essere fatta con cautela. Sono necessari ulteriori studi per comprendere meglio il quadro clinico e trovare nuovi approcci terapeutici.
Messaggi da portare a casa
- La CPVT si verifica quasi esclusivamente nell’infanzia e nell’adolescenza in cuori strutturalmente normali e ha un alto tasso di mortalità se non viene trattata.
- Le terapie principali sono l’adeguamento dello stile di vita e i beta-bloccanti.
- L’impianto di ICD deve essere eseguito solo nei pazienti con morte cardiaca improvvisa sopravvissuta, sincope o tachicardia ventricolare polimorfa persistente nonostante la terapia farmacologica massima o dopo la valutazione della denervazione simpatica cardiaca sinistra.
- L’erogazione dello shock da parte di un ICD ha un rischio elevato di proaritmia, quindi i tempi di rilevamento lunghi e le frequenze di intervento elevate devono essere programmati nell’ICD.
Letteratura:
- Reid DS, et al: Tachicardia bidirezionale in un bambino. Uno studio che utilizza l’elettrografia del fascio di His. Br Heart J. 1975; 37:339-344.
- Leenhardt A, et al: Tachicardia ventricolare polimorfa catecolaminergica nei bambini. Un follow-up di 7 anni di 21 pazienti. Circolazione. 1995; 91: 1512-1519.
- Priori SG, et al: Caratterizzazione clinica e molecolare dei pazienti con tachicardia ventricolare polimorfa catecolaminergica. Circolazione. 2002; 106: 69-74.
- Leenhardt Antoine, Denjoy Isabelle, Guicheney Pascale. Tachicardia ventricolare polimorfa catecolaminergica. Aritmo Circolare Elettrofisiologia. 2012; 5: 1044-1052.
- Pérez-Riera AR, et al: Tachicardia ventricolare polimorfa catecolaminergica, un aggiornamento. Ann Noninvasive Electrocardiol Off J Int Soc Holter Noninvasive Electrocardiol Inc. 2018;23: e12512.
- Dichiarazione di consenso degli esperti HRS/EHRA/APHRS sulla diagnosi e la gestione dei pazienti con sindromi di aritmia primaria ereditaria. 2013; 69.
- van der Werf C, Wilde AAM: Tachicardia ventricolare polimorfa catecolaminergica: dal banco al letto del paziente. Cuore. 2013; 99: 497-504.
- Svendsen JH, Geelen P, Comitato d’iniziativa scientifica EHRA: Screening e gestione di possibili sindromi aritmogene (canalopatie/malattie dei canali ionici). Eur Eur Pacing Arrhythm Card Electrophysiol J Gruppi di lavoro Card Pacing Arrhythm Card Cell Electrophysiol Eur Soc Cardiol. 2010; 12: 741-742.
- Sy RW, et al: Caratterizzazione dell’aritmia ed esiti a lungo termine nella tachicardia ventricolare polimorfa catecolaminergica. Ritmo cardiaco. 2011; 8:864-871.
- Sumitomo N, et al.: Tachicardia ventricolare polimorfa catecolaminergica: caratteristiche elettrocardiografiche e strategie terapeutiche ottimali per prevenire la morte improvvisa. Cuore. 2003;89: 66-70.
- Tester DJ, et al: Annegamenti inspiegabili e canalopatie cardiache: una serie di autopsie molecolari. Mayo Clin Proc. 2011; 86: 941-947.
- Sy RW, et al: Derivazione e validazione di un semplice algoritmo basato sull’esercizio fisico per la previsione di test genetici nei parenti di probandi LQTS. Circolazione. 2011; 124: 2187-2194.
- Inoue YY, et al.: Risposte diverse all’esercizio fisico tra la sindrome di Andersen-Tawil e la tachicardia ventricolare polimorfa catecolaminergica. Eur Eur Pacing Arrhythm Card Electrophysiol J Gruppi di lavoro Card Pacing Arrhythm Card Cell Electrophysiol Eur Soc Cardiol. 2018; 20: 1675-1682.
- Berte B, et al: Tachicardia ventricolare bidirezionale nella miocardite fulminante. Eur Eur Pacing Arrhythm Card Electrophysiol J Gruppi di lavoro Card Pacing Arrhythm Card Cell Electrophysiol Eur Soc Cardiol 2008; 10: 767-768.
- Valent S, Kelly P: Immagini nella medicina clinica. Tachicardia ventricolare bidirezionale indotta dalla digossina. N Engl J Med. 1997; 336: 550.
- Hayashi M, et al.: Incidenza e fattori di rischio di eventi aritmici nella tachicardia ventricolare polimorfa catecolaminergica. Circolazione. 2009; 119: 2426-2434.
- Ostby SA, et al: Partecipazione sportiva agonistica in pazienti con tachicardia ventricolare polimorfa catecolaminergica: la prima esperienza di un singolo centro. JACC Clin Electrophysiol. 2016; 2: 253-262.
- Leren IS, et al: Il Nadololo riduce l’incidenza e la gravità delle aritmie ventricolari durante il test da sforzo rispetto ai β1-bloccanti selettivi nei pazienti con tachicardia ventricolare polimorfa catecolaminergica. Ritmo cardiaco. 2016; 13: 433-440.
- van der Werf C, Zwinderman AH, Wilde AAM: Approccio terapeutico per i pazienti con tachicardia ventricolare polimorfa catecolaminergica: stato dell’arte e sviluppi futuri. Eur Eur Pacing Arrhythm Card Electrophysiol J Gruppi di lavoro Card Pacing Arrhythm Card Cell Electrophysiol Eur Soc Cardiol. 2012; 14:175-183.
- Zhou Q, et al: il carvedilolo e i suoi nuovi analoghi sopprimono il rilascio di Ca2+ indotto dal sovraccarico aritmogeno del magazzino. Nat Med. 2011; 17: 1003-1009.
- van der Werf C, et al: la terapia con flecainide riduce le aritmie ventricolari indotte dall’esercizio fisico nei pazienti con tachicardia ventricolare polimorfa catecolaminergica. J Am Coll Cardiol. 2011; 57: 2244-2254.
- Watanabe H, et al: La flecainide previene la tachicardia ventricolare polimorfa catecolaminergica nei topi e negli esseri umani. Nat Med. 2009; 15: 380-383.
- Rosso R, et al.: Calcio-antagonisti e beta-bloccanti rispetto ai soli beta-bloccanti per prevenire le aritmie indotte dall’esercizio fisico nella tachicardia ventricolare polimorfa catecolaminergica. Ritmo cardiaco. 2007; 4: 1149-1154.
- Swan H, et al: l’antagonismo dei canali del calcio riduce le aritmie ventricolari indotte dall’esercizio fisico nei pazienti con tachicardia ventricolare polimorfa catecolaminergica con mutazioni RyR2. J Cardiovasc Electrophysiol. 2005; 16: 162-166.
- Hwang HS, et al: L’inibizione dei canali di rilascio del Ca2+ cardiaco (RyR2) determina l’efficacia dei farmaci antiaritmici di classe I nella tachicardia ventricolare polimorfa catecolaminergica. Circ Arrhythm Electrophysiol. 2011; 4: 128-135.
- Jung CB, et al: Il dantrolene salva il difetto aritmogenico RYR2 in un modello di cellule staminali paziente-specifiche di tachicardia ventricolare polimorfa catecolaminergica. EMBO Mol Med. 2012; 4: 180-191.
- Odero A, et al: Denervazione del simpatico cardiaco sinistro per la prevenzione di aritmie pericolose per la vita: l’approccio chirurgico sovraclaveare alla simpaticotomia cervicotoracica. Ritmo cardiaco. 2010; 7: 1161-1165.
- Wilde AAM, et al: Denervazione simpatica cardiaca sinistra per la tachicardia ventricolare polimorfa catecolaminergica. N Engl J Med. 2008; 358: 2024-2029.
- Mohamed U, et al: Morte cardiaca improvvisa nonostante un cardioverter-defibrillatore impiantabile in una giovane donna con tachicardia ventricolare catecolaminergica. Ritmo cardiaco. 2006; 3: 1486-1489.
CARDIOVASC 2019; 18(2): 12-15