Se la formazione di nuove cellule del sangue nel midollo osseo è disturbata, la causa può essere una rara forma di cancro cronico del sangue. L’iperproliferazione delle tre serie di cellule nel midollo osseo causa eritrocitosi, trombocitosi e leucocitosi nella policitemia vera. Il risultato è, tra l’altro, un aumento significativo dei livelli di ematocrito e quindi anche del rischio di eventi tromboembolici.
La policitemia vera (PV) è una neoplasia mieloproliferativa cronica molto rara ed è caratterizzata da un aumento dell’ematopoiesi. La maggior parte dei pazienti con PV presenta una mutazione nel gene della tirosin-chinasi JAK2 [1]. Ne consegue un aumento della proliferazione cellulare e della produzione di citochine proinfiammatorie. La sovrapproduzione di eritrociti e il conseguente aumento dell’ematocrito aumentano la viscosità del sangue. In questo modo, si favorisce l’insorgenza di tromboembolismo: il 45% di tutti i decessi in PV è dovuto a complicazioni tromboemboliche [2]. Tuttavia, la prognosi è generalmente favorevole. L’età mediana alla diagnosi è di 65 anni.
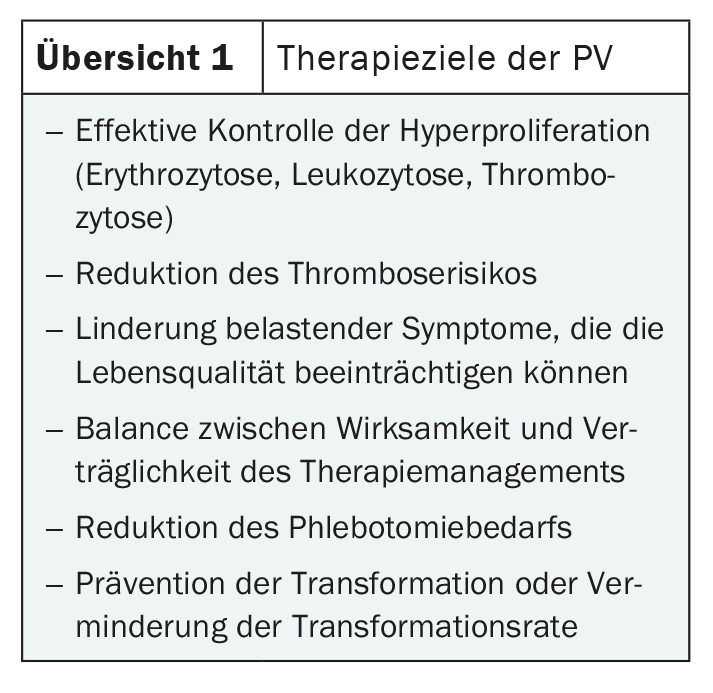
Sebbene i sintomi siano vari, in genere sono piuttosto aspecifici. Pertanto, la diagnosi viene spesso fatta solo per caso. I possibili sintomi includono mal di testa, disturbi visivi, affaticamento e prurito, oltre a dolore osseo e dolore nella parte superiore dell’addome. Spesso sono causati dalla splenomegalia tipica della FV [1,2]. L’aumento della massa delle cellule del sangue può causare disturbi circolatori che possono portare a gravi trombosi venose e arteriose, come embolia polmonare, apoplessia o infarto del miocardio. Sono quindi indicati una diagnosi precoce e un trattamento efficace (Panoramica 1) [3].
Nella fase cronica, che di solito dura anni, le caratteristiche cliniche dell’aumento della mieloproliferazione sono al centro dell’attenzione. Le complicazioni più frequenti e potenzialmente minacciose sono le tromboembolie arteriose o venose, che si verificano fino al 40% dei pazienti. Nella fase avanzata della malattia, il problema principale risiede nella cosiddetta fase ‘ritardata’. Questa è caratterizzata da una diminuzione dell’eritrocitosi e da un aumento della splenomegalia, in combinazione con la fibrosi del midollo osseo, che può essere seguita dalla trasformazione in mielofibrosi (secondaria) post-PV e/o in leucemia acuta [1]. Il tasso complessivo di post-PV-MF è di circa il 15% dopo un periodo di osservazione mediano di 10 anni e del 50% dopo 20 anni.
Trattamento adattato al rischio
Poiché la prevenzione della tromboembolia è fondamentale, la flebotomia è spesso considerata il trattamento di scelta. Questo può abbassare l’ematocrito (Hct) al di sotto del 45% e ridurre l’iperviscosità del sangue. Gli studi hanno dimostrato che impostare l’Hct al di sotto del 45% può ridurre il tasso di morte cardiovascolare in PV fino a un quarto [4]. Tuttavia, la flebotomia è molto faticosa. Pertanto, inizialmente deve essere iniziato anche il trattamento con acido acetilsalicilico (ASA) a basso dosaggio. La raccomandazione di trattamento si basa quindi sul punteggio di rischio. Si può ipotizzare un rischio basso per i pazienti più giovani, di età inferiore ai 60 anni, che non hanno avuto in precedenza una trombosi. Attualmente si sta discutendo se il trattamento di riduzione delle citochine debba essere preso in considerazione anche per loro, in determinate condizioni.

Tuttavia, la maggior parte dei pazienti con FV è comunque ad alto rischio. In questi casi, è indicato l’avvio di una terapia citoriduttiva. L’idrossiurea (HU) o l’interferone alfa (INF) sono raccomandati per il trattamento primario [5]. Tuttavia, l’HU in particolare non è adatto a tutti i pazienti e può causare gravi effetti collaterali (Panoramica 2) [6]. Per i pazienti più giovani che desiderano avere figli, è più probabile che si ricorra all’INF. Se la terapia di prima linea non è tollerata o i sintomi clinici non regrediscono sufficientemente, il trattamento deve essere cambiato. L’inibitore di JAK2 ruxolitinib ha dimostrato negli studi di controllare l’aumento della mieloproliferazione e di essere ben tollerato [1]. Sono scomparsi anche molti sintomi associati alla FV, come l’affaticamento e il prurito. Inoltre, la maggior parte dei pazienti ha sperimentato l’effetto molto rapidamente, entro le prime quattro settimane. Il busulfano può essere utilizzato come terapia alternativa nei pazienti in età avanzata. In questo caso, tuttavia, il potenziale leucemogeno è sempre in discussione, motivo per cui la sostanza dovrebbe essere utilizzata solo con moderazione. L’anagrelide può essere considerata come partner di combinazione con, ad esempio, HU o INF. Questo è finalizzato esclusivamente a ridurre la produzione di piastrine e può fungere da complemento se non si ottengono risultati soddisfacenti con le altre sostanze da sole.
Letteratura:
- www.onkopedia.com/de/onkopedia/guidelines/polycythaemia-vera-pv/@@guideline/html/index.html (ultima chiamata il 23/04/2024).
- Vannucchi AM, et al.: N Engl J Med 2015; 372: 426–435.
- Stein BL, Moliterno AR, Tiu RV: Polycythemia vera disease burden: contributing factors, impact on quality of life, andemerging treatment options. Ann Hematol 2014; 93: 1965–1976.
- Marchioli R, Finazzi G, Specchia G, et et al.: Cardiovascular events and intensity of treatment in polycythemia vera. N Engl J Med 2013; 368: 22–33.
- Barbui T, Tefferi A, Vannucchi AM, et al.: Philadelphia chromosome-negative classical myeloproliferative neoplasms: revised management recommendations from European Leukemia Net. Leukemia 2018; 32: 1057–1069.
- Barosi G, Birgegard G, Finazzi G, et al.: A unified definition of clinical resistance and intolerance to hydroxycarbamide in polycythaemia vera and primary myelofibrosis: results of a European LeukemiaNet (ELN) consensus process. Br J Haematol 2010; 148: 961–963.
InFo ONKOLOGIE & HÄMATOLOGIE 2024; 12(2): 38