Una casalinga di 54 anni ha avuto un episodio acuto di angioedema della lingua e della parete faringea posteriore e ha richiesto un trattamento di emergenza nel reparto di terapia intensiva (Fig. 1). Poiché i gonfiori si sono verificati 30 minuti dopo aver mangiato spaghetti al pomodoro e vongole, è stato sospettato un evento allergico e la paziente è stata indirizzata per un chiarimento allergologico. Il referto medico parlava di una lieve pancitopenia preesistente.
L’anamnesi allergologica era improduttiva per quanto riguarda le malattie atopiche preesistenti; in particolare, non c’erano prove di precedenti reazioni allergiche o di intolleranza in relazione agli alimenti. Di conseguenza, i prick test (screening dell’atopia, panoramica degli alimenti, comprese le cozze) e le determinazioni delle IgE sono risultati negativi. Prima dell’episodio, la paziente non assumeva preparati contenenti ASA o farmaci antinfiammatori non steroidei, né era in terapia con ACE-inibitori o antagonisti dell’angiotensina (AT) II (sartani) [1, 2].
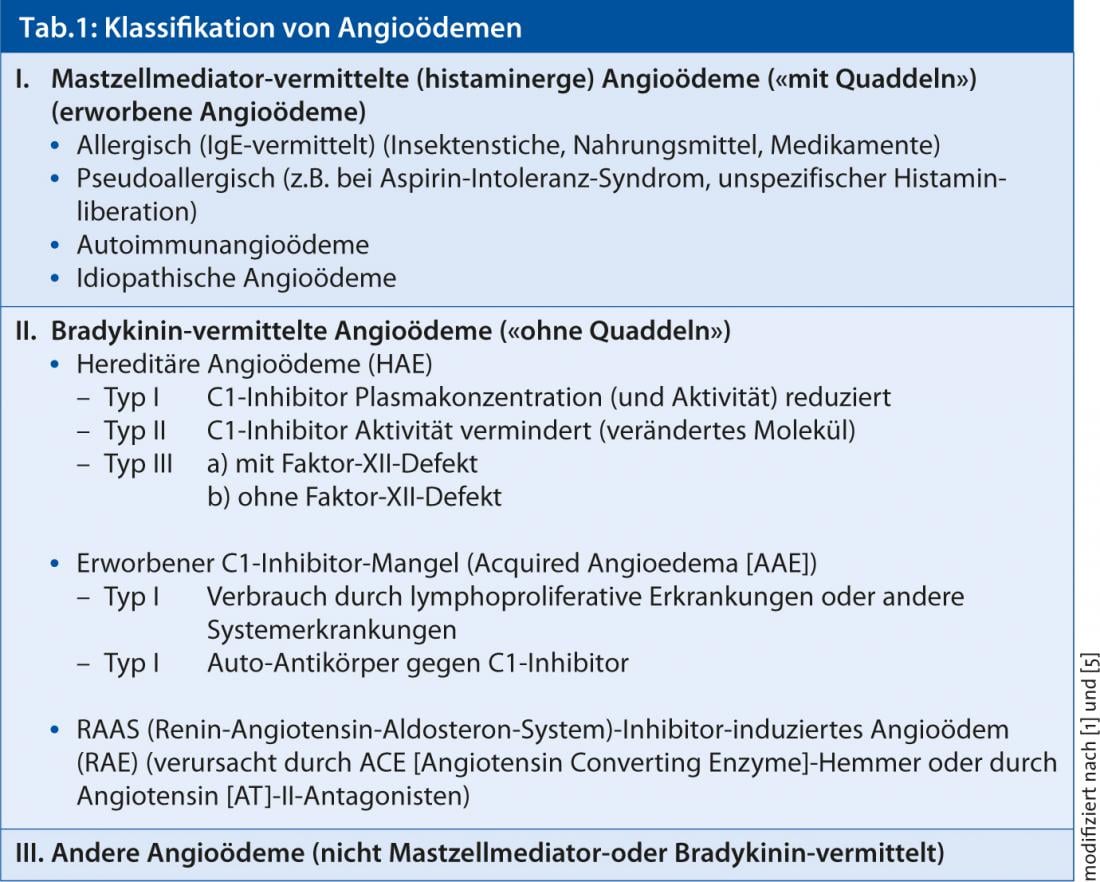
Nella considerazione diagnostica differenziale dell’angioedema (Tabella 1) , in assenza di indizi anamnestici per i fattori scatenanti, il dolore addominale occasionalmente lamentato è stato interpretato in relazione alla particolare localizzazione dell’episodio di angioedema nel senso di un angioedema ereditario ed è stato esaminato il profilo del complemento.
Sia la proteina inibitrice di C1 (<0,03 g/l; intervallo normale 0,2-0,36) che i livelli di C4 (<0,01 g/l; intervallo normale 0,1-0,4) non erano misurabili. Allo stesso modo, la funzione di C1 inibitore era molto bassa: <18% (norma 70-135%; Laboratorio Immunologico, USZ).
Con la diagnosi di lavoro di angioedema ereditario (tipo I), è stata iniziata una terapia con l’androgeno attenuato danazol (Danatrol), inizialmente 40 mg/die per quindici giorni, poi 200 mg/die per altre due settimane, per la profilassi delle crisi. Un controllo dei parametri di coagulazione ha rivelato la presenza di anticorpi antifosfolipidi (anticardiolipina -IgM 520 UI/l; norma <15 UI/l); la sierologia per gli autoanticorpi antinucleari e anti-DNA era negativa, escludendo così il lupus eritematoso sistemico (LES). La diagnosi originale di angioedema ereditario è stata rivista in “angioedema acquisito [AAE] con carenza di C1 inibitore in associazione alla sindrome antifosfolipidica (APS)”.
La diagnosi di AAE è stata successivamente confermata con la determinazione dei livelli di C1q e C1r diminuiti due volte (C1q 0,014 g/l; norma: 0,0460-1116 g/l e C1r 37%; norma: 75-125%). Infine, una biopsia del midollo osseo ha portato alla diagnosi di linfoma non-Hodgkin a cellule B (NHL, IgG κ).
Diagnosi
Angioedema acquisito (AAE) nella carenza acquisita di C1 inibitore (tipo I) dovuta al consumo/presenza di un inibitore in un linfoma non Hodgkin.
Discussione
In base ai mediatori coinvolti, l’angioedema viene ora suddiviso patogeneticamente in mediato dai mastociti o istaminergico (spesso accompagnato da orticaria) e mediato dalla bradichinina (senza focolai) (Tabella 1) [1, 5]. Tra questi ultimi, oltre all’angioedema ereditario (HAE) (tipi II e III) dovuto alla carenza di C1 inibitore e all’angioedema indotto da inibitori del sistema renina-angiotensina-aldosterone (RAAS) causato da ACE inibitori o antagonisti dell’AT-II, esiste un’ulteriore forma estremamente rara di angioedema, caratterizzata anche da una carenza di C1 inibitore dell’esterasi (C1-INH) [4]. Tuttavia, la carenza in questo caso non è causata da un cambiamento nei geni (fattori ereditari), ma da alcune malattie o processi “acquisiti” nell’organismo che consumano o inattivano (o inibiscono) sempre più il C1-INH. La conseguenza: c’è troppo poco (funzionale) C1-INH disponibile per rallentare la formazione eccessiva di bradichinina. I livelli di bradichinina aumentano e si può sviluppare un gonfiore sulla pelle e sulle membrane mucose. A differenza dell’angioedema ereditario, questa rara forma di angioedema mediato da bradichinina si manifesta solitamente dopo i 30 anni e non colpisce altri membri della famiglia. L’angioedema acquisito può essere il concomitante di un’altra malattia di base, che deve essere comunque chiarita da un medico.
Nel caso in questione, inizialmente è stato decisivo determinare i fattori di complemento C4 e C1 inibitore per escludere l’angioedema istaminergico, ad esempio in caso di sospetta allergia alimentare. Un’ulteriore determinazione dei primi componenti del complemento C1q e C1r è servita a differenziare tra un HAE e un AAE. La malattia di base nell’AAE doveva essere ulteriormente indagata dal punto di vista sierologico (esclusione di malattie autoimmuni, come il LES, ecc.) e dalla biopsia del midollo osseo [6]. La pancitopenia potrebbe essere spiegata nel contesto del coinvolgimento del midollo osseo del linfoma non-Hodgkin.
Conclusione
Nella maggior parte dei casi, l’angioedema è dovuto a cause allergiche. Tuttavia, esistono anche angioedemi non causati da allergie, che si verificano in modo comparativamente molto raro e il cui background medico spesso rimane inosservato per un lungo periodo di tempo. Poiché i diversi tipi di angioedema richiedono trattamenti diversi, occorre innanzitutto identificare la causa del gonfiore.
Fondamentalmente, si distinguono le seguenti forme di angioedema:
- Angioedema mediato da istamina (questo include sia l’allergia che l’angioedema indotto da farmaci).
- Angioedema mediato da bradichinina o angioedema ereditario (HAE) causato da un difetto o da una carenza di C1 inibitore.
Questo caso clinico ha dimostrato che la carenza di C1 inibitore non solo è ereditaria, ma raramente può anche essere acquisita, nel qual caso si parla di angioedema acquisito (AAE). Il fattore decisivo per la diagnosi è l’esame di laboratorio. L’esecuzione di prick test e la determinazione degli inibitori di C1 e C4 consentono di escludere l’angioedema istaminergico come primo passo. Misurando i livelli dei singoli valori ematici C1q e C1r, si può fare una distinzione tra HAE e AAE in un ulteriore passo. Mentre il primo gonfiore
Mentre gli attacchi di HAE si verificano sempre prima dei 30 anni, la rara AAE compare solitamente dopo questa età. Un AAE può essere un indicatore di una malattia sottostante e non deve essere ignorato.
Letteratura:
- Wüthrich B: Angioedema. Raramente allergico. Parte prima: Classificazione, fisiopatologia, diagnosi. Switzerland Med Forum 2012; 12 (7): 138-143.
- Wüthrich B: Angioedema. Raramente allergico. Parte seconda: Terapia. Switzerland Med Forum 2012;12 (8): 175-178.).
- Wüthrich B: È presente l’edema di Quincke? Diagnosi differenziale con l’angioedema. DERMATOLOGIE PRAXIS 2012; 1: 20.
- Wüthrich B: Qual è la sua diagnosi? (Quiz). DERMATOLOGIE PRAXIS 2011;3: 38 e 42.
- Maurer M, Magerl M: JDDG 2010; 8/9: 663-672.
- Beretta KR, et al: Schweiz Med Wschr 1991; 121: 943-947.