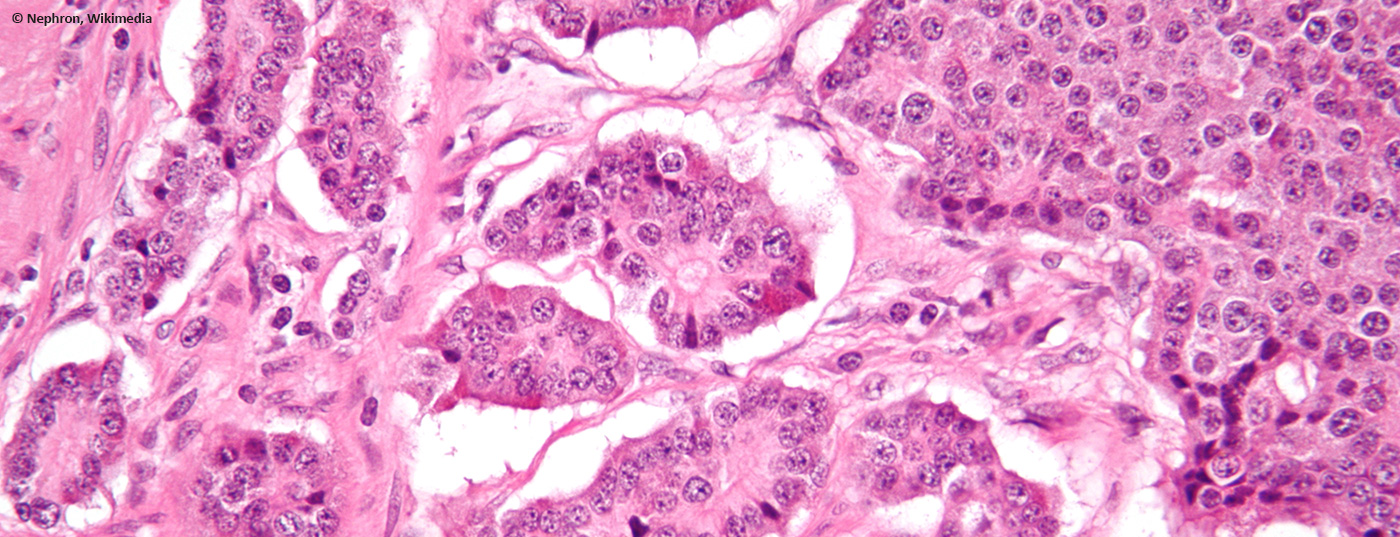
Le neoplasie neuroendocrine di solito si verificano sporadicamente e spesso hanno origine nello spazio gastroenteropancreatico. Di solito vengono diagnosticati incidentalmente, perché spesso non sono funzionali. Pertanto, nella maggior parte dei casi si tratta di una malattia tumorale già avanzata, spesso metastatizzata.
Il termine ‘neuroendocrino’ si riferisce a cellule con caratteristiche istologiche di tessuto ghiandolare, che di solito sono presenti in modo diffuso in vari organi del corpo e possono secernere ormoni. A seconda della funzione e della localizzazione, vengono prodotte ammine (ad esempio, catecolamine) o peptidi (ad esempio, somatostatina), che regolano l’attività del tessuto circostante. Dal punto di vista embriologico, sono state a lungo considerate parte della cresta neurale, ma oggi sono classificate come parte dell’endoderma, proprio come le cellule esocrine del tratto gastrointestinale. Storicamente, queste cellule sono state raggruppate in un sistema neuroendocrino diffuso (in passato chiamato anche APUD (“amine precursor uptake and decarboxylation”)). Tuttavia, poiché questi termini non hanno alcun significato istopatologico o clinico rilevante, non dovrebbero più essere utilizzati.
Se queste cellule degenerano, vengono chiamate neoplasie neuroendocrine (NEN). Questo termine ombrello comprende sia i tumori neuroendocrini (NET) a lenta proliferazione e i carcinoidi del polmone, sia i carcinomi neuroendocrini (NEC), significativamente più aggressivi e prognosticamente sfavorevoli. Tutte le raccomandazioni contenute in questo articolo si baseranno quindi su questa classificazione.
Le neoplasie neuroendocrine si verificano per lo più sporadicamente, con un’incidenza stimata di circa 5/100.000 abitanti. Un cluster familiare può essere osservato nella neoplasia endocrina multipla (MEN) I e II, nonché in malattie rare come la sindrome di von Hippel-Lindau o la neurofibromatosi di tipo 1 (Recklinghausen). La maggior parte delle NEN ha origine dallo spazio gastroenteropancreatico (GEP-NEN), circa il 25-30% ha origine dal polmone [1].
Sintomatologia
Sebbene la produzione di ormoni sia una caratteristica tipica delle neoplasie neuroendocrine, i cosiddetti tumori funzionali – cioè i tumori la cui produzione e secrezione ormonale provoca sintomi – sono rari. I disturbi causati dai tumori funzionali dipendono dalla rispettiva secrezione di sostanze bioattive. Gli insulinomi o i gastrinomi, che rappresentano la maggior parte di queste NEN funzionali, portano, ad esempio, a ipoglicemia grave, potenzialmente pericolosa per la vita, o a ulcerazioni gastriche multiple (sindrome di Zollinger-Ellison). Un altro quadro clinico tipico è la sindrome da carcinoide, in cui l’eccessiva secrezione di serotonina può portare a una grave diarrea acquosa, a sintomi di vampate e a broncospasmo. La fibrosi endocardica può verificarsi come complicazione tardiva, che può portare a un’insufficienza cardiaca con conseguenze fatali (sindrome di Hedinger).
Tuttavia, fino al 90% di tutte le NEN non sono funzionali e vengono diagnosticate come reperti accidentali o nel contesto di disturbi non specifici. Nella maggior parte dei casi, a causa del decorso indolente, si tratta di una malattia tumorale avanzata e già metastatizzata.
Diagnostica
Per confermare la diagnosi è sempre necessaria una biopsia. Il work-up istopatologico deve includere la determinazione dei marcatori tipici (sinaptofisina, recettori della somatostatina [SSTR]) e necessariamente il tasso di proliferazione (Ki-67 o MIB-1) o il tasso di mitosi, poiché questi sono di notevole importanza sia prognostica che terapeutica. Nel caso di una sospetta sindrome carcinoide, la determinazione dell’acido 5-idrossi-indoleacetico (5-HIESS, un prodotto di degradazione della serotonina) nelle urine delle 24 ore è diagnostica. Altri valori di laboratorio a digiuno, come la cromogranina A (CgA) e, se necessario, l’enolasi neurone-specifica (NSE), possono essere utilizzati come marcatori tumorali. A seconda della clinica, vengono utilizzati altri test (ad esempio, il test del digiuno per l’insulinoma). Come prospettiva futura, va menzionato anche un nuovo metodo: il cosiddetto NETest [2]. Si tratta di una PCR diagnostica da sangue intero (“biopsia liquida”), con la quale vengono esaminati 51 filamenti di mRNA specifici del NET. Il valore predittivo positivo per la diagnosi di neoplasia neuroendocrina è superiore al 90%, così come la sensibilità e la specificità. I primi rapporti hanno dimostrato la sua utilità sia per la diagnosi che per il monitoraggio della risposta al trattamento. Al momento, tuttavia, questo NETest non è ancora ampiamente disponibile.
Imaging
Oltre alla consueta diagnostica per immagini, l’imaging nucleare funzionale svolge un ruolo cruciale, sia nella stadiazione che nella selezione della terapia. In primo luogo, va menzionata la PET/CT con 68Ga-DOTATATE, che negli ultimi anni ha sostituito costantemente la scintigrafia con octreotide, meno precisa e più complessa [3]. Grazie a questo esame PET significativamente più sensibile, non è raro che si verifichi un upstaging attraverso il rilevamento di metastasi non visibili nella tomografia computerizzata. Inoltre, può essere utilizzato per rilevare l’espressione di SSTR2, spesso presente sulla superficie cellulare del NET, e quindi valutare il possibile uso della terapia con radionuclidi (PRRT, vedi sotto). Se l’indice di proliferazione è elevato o se l’espressione di SSTR2 è assente o eterogenea, è utile la prestazione aggiuntiva della FDG-PET/CT [4], perché le lesioni FDG-avide indicano un comportamento più aggressivo e devono essere esaminate ulteriormente a livello bioptico, quando possibile.
Nuova classificazione OMS
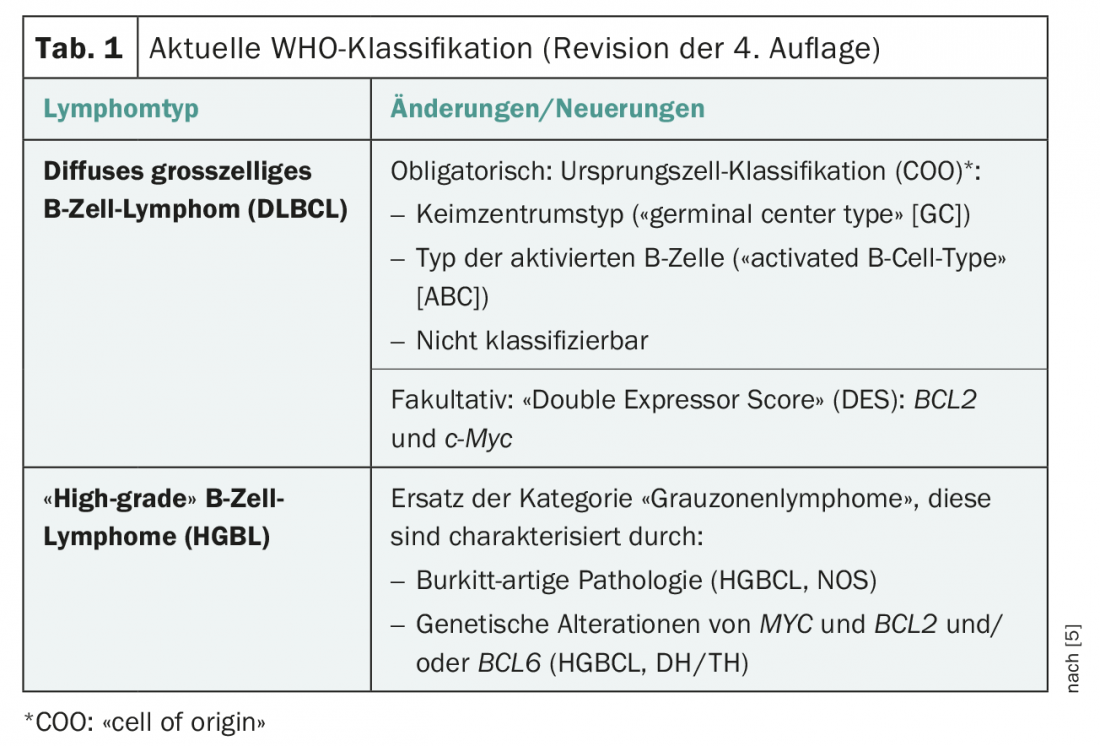
Negli ultimi decenni, ci sono stati cambiamenti significativi nella classificazione delle neoplasie neuroendocrine, grazie a nuovi risultati istopatologici e clinici (Tab. 1) . L’attuale classificazione dell’OMS (2017) ha eliminato, in particolare per le NEN del pancreas (nuove: panNEN), le discrepanze istopatologiche (alto indice di proliferazione).
ma con un basso tasso di mitosi) e le caratteristiche cliniche (prognosi) sono state prese in considerazione. Ne consegue la suddivisione in NET G1, NET G2, NET G3 e NEC G3. Dalla classificazione del 2010, il termine “carcinoide” deve essere utilizzato solo per il NET polmonare (Tab. 2).

Trattamento del NET non metastatico e follow-up
L’unica opzione potenzialmente curativa per le neoplasie neuroendocrine è l’asportazione completa del tumore. Nel tratto gastrointestinale, questo può essere fatto per via endoscopica, a seconda delle dimensioni (fino a 2 cm) e del sito. Tuttavia, l’indicazione alla resezione chirurgica deve essere fatta a bassa soglia se c’è evidenza di infiltrazione della muscolatura propria (T2) o di metastasi linfonodali [5]. Le reti dell’appendice sono una situazione speciale. Nella stragrande maggioranza dei casi, si tratta di un reperto incidentale dopo un’appendicectomia. La decisione di eseguire un’emicolectomia destra supplementare con linfoadenectomia dipende dalle dimensioni del tumore (>2 cm), dalla profondità di penetrazione (>3 mm) nel mesoappendice e dall’invasione vascolare (V1) o linfatica (L1) [6].
A differenza di altre entità tumorali, non esiste alcuna indicazione per una terapia adiuvante nei tumori neuroendocrini. Si raccomanda un follow-up regolare con endoscopie annuali per il NET dello stomaco o del retto. Nel caso di NET localmente avanzati, ma completamente rimossi chirurgicamente, la diagnostica per immagini annuale (di solito TAC addome toracico con mezzo di contrasto i.v.) deve essere eseguita per tutta la vita.
Opzione terapeutica per la NEN metastatica
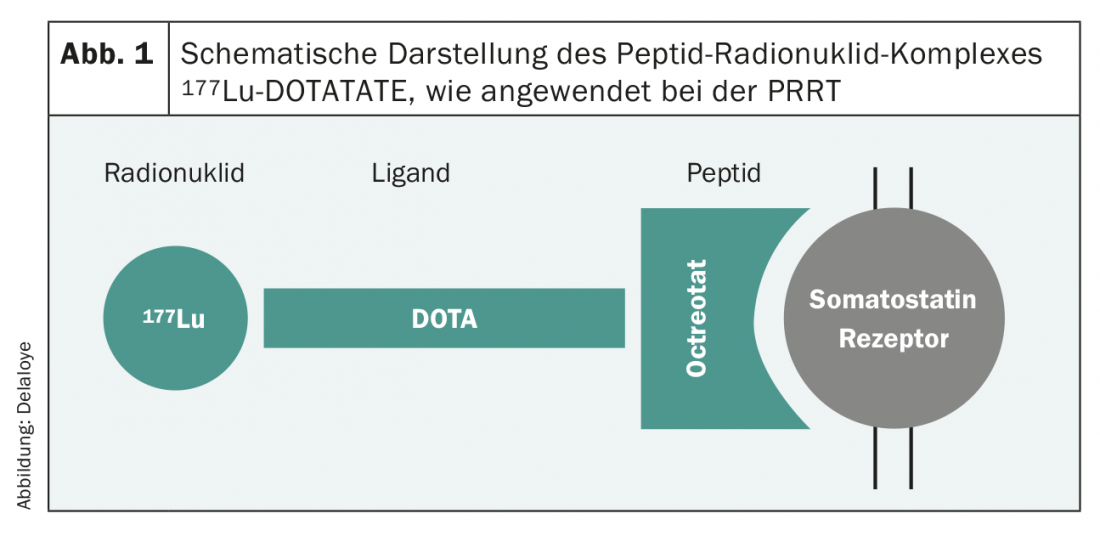
I nuovi sviluppi dell’ultimo decennio hanno migliorato drasticamente il panorama terapeutico e quindi la sopravvivenza dei pazienti. Il numero di studi di fase III rilevanti per la pratica si è sviluppato in modo quasi esponenziale e le raccomandazioni delle società professionali si basano su prove sempre più solide. Fondamentalmente, le opzioni di trattamento per i tumori G1 e G2 sono molto simili. In questa popolazione di pazienti, occorre prestare particolare attenzione agli effetti collaterali della terapia, poiché si tratta per lo più di pazienti asintomatici. Nei tumori che esprimono SSTR2, numerosi studi hanno stabilito l’uso in prima linea degli analoghi della somatostatina [7,8], anche nel tumore non funzionale. Questa terapia, che di solito è ben tollerata, spesso porta a un buon controllo del tumore e quindi a un prolungamento della sopravvivenza libera da progressione. Un’altra opzione è la terapia con radionuclidi peptidici (PRRT). Una procedura in cui una sostanza radioattiva (90 Ittrio o 177Lutezio), legata a un peptide affine al recettore della somatostatina (Octreotide/Octreotate) (Fig. 1), viene somministrata per via endovenosa come infusione e consente l’irradiazione mirata delle cellule tumorali SSTR2-positive. Questo trattamento viene solitamente applicato quattro volte di seguito, ogni volta a un intervallo di otto settimane. Si tratta anche di una terapia che di solito è molto ben tollerata e i cui risultati sono stati dimostrati qualche anno fa in uno studio di fase III su larga scala [9].
A livello molecolare, l’uso di everolimus come inibitore di mTOR è ormai consolidato. Il meccanismo di proliferazione cellulare attorno alla via PI3K-mTOR svolge un ruolo importante nelle neoplasie neuroendocrine, come in altri tumori. Pertanto, è possibile ottenere un buon controllo del tumore con everolimus [10], ma questa terapia è associata a un numero significativamente maggiore di effetti collaterali.
La chemioterapia viene utilizzata anche per i G2-NET e G3-NET più proliferanti. Uno studio di fase II ha dimostrato l’attività antitumorale di capecitabina e temozolomide (CapTem) in questa coorte di pazienti [11]. Sebbene manchino ancora dati di fase III, questa terapia ha dato prova di sé nella pratica e si è rapidamente affermata come alternativa alla precedente combinazione di streptozotocina e 5-fluorouracile (STZ+5FU). Anche il profilo degli effetti collaterali è favorevole e la gestione è semplice.
Le neoplasie neuroendocrine di grado superiore (cioè NEC G3) vengono trattate con una combinazione di platino ed etoposide analoga alla terapia per il carcinoma bronchiale a piccole cellule. Le nuove terapie con gli inibitori del checkpoint hanno mostrato finora uno scarso successo e non possono essere generalmente raccomandate. Devono essere ulteriormente studiati in studi clinici.
Messaggi da portare a casa
- Le cellule con caratteristiche istologiche di tessuto ghiandolare, che di solito sono presenti in modo diffuso in vari organi del corpo e possono secernere ormoni, sono definite “neuroendocrine”. Se degenerano, si chiamano neoplasie neuroendocrine (NEN).
- La maggior parte delle NEN ha origine dallo spazio gastroenteropancreatico, mentre circa il 25-30% ha origine dai polmoni.
- I tumori funzionali, come gli insulinomi o i gastrinomi, sono rari, ma possono portare a ipoglicemia grave, potenzialmente pericolosa per la vita, o a ulcerazioni gastriche multiple.
- Di norma, tuttavia, le NEN non sono funzionali e vengono diagnosticate come reperti accidentali o nel contesto di disturbi non specifici.
- L’unica opzione potenzialmente curativa per le neoplasie neuroendocrine non metastatiche è l’asportazione completa del tumore. A differenza di altre entità tumorali, non esiste alcuna indicazione per una terapia adiuvante nei tumori neuroendocrini.
- Le opzioni di trattamento della NEN metastatica sono migliorate notevolmente. Per i tumori che esprimono SSTR2, è ormai consolidato l’uso degli analoghi della somatostatina. Le neoplasie neuroendocrine di grado superiore vengono trattate con una combinazione di platino ed etoposide analoga alla terapia per il carcinoma bronchiale a piccole cellule.
Letteratura:
- Cives M, et al: CA Cancer J Clin 2018; 68: 471-487.
- Modlin IM, et al: Endocr Relat Cancer 2014; 21(4): 615-628.
- Deppen A, et al: J Nucl Med 2016; 57(6): 872-878
- Kayani I, et al.: Cancer 2008; 112(11): 2447-2455
- Delle Fave G, et al: Neuroendocrinologia 2016; 103(2): 119-124
- Pape UF, et al: Neuroendocrinologia 2016; 103(2): 144-152
- Rinke A, et al: J Clin Oncol 2009; 27(28): 4656-4663.
- Caplin ME, et al: N Engl J Med 2014; 371(3): 224-233.
- Strosberg J, et al: N Engl J Med 2017; 376(2): 125-135
- Yao J, et al: Lancet 2016; 387: 968-977
- Strosberg JR, et al: Cancer 2011; 117(2): 268-275
InFo ONcOLOGIA & EMATOLOGIA 2019; 7(1): 16-18.