Típico da amiloidose cardíaca é o desenvolvimento de insuficiência cardíaca com fração de ejeção preservada (HFpEF). Novos procedimentos de diagnóstico e opções terapêuticas estão agora a lançar uma luz diferente sobre esta doença rara, até agora praticamente incurável.
Com a introdução de novos procedimentos de diagnóstico e opções terapêuticas para o que tem sido uma doença rara praticamente incurável, o interesse pela amiloidose está a crescer rapidamente. O envolvimento cardíaco (amiloidose cardíaca) leva geralmente a uma insuficiência cardíaca grave com todas as suas consequências clínicas. O objectivo deste artigo é dar uma breve visão geral do diagnóstico moderno e da terapia da amiloidose cardíaca.
Origem
Na amiloidose sistémica, existem depósitos de proteínas anormalmente dobradas e agregadas no tecido. Dependendo da infestação de órgãos, apresentam-se as correspondentes perturbações funcionais e doenças. Se os depósitos estiverem no coração (no interstício entre as células do miocárdio), isto leva a doença cardíaca com o quadro clínico de insuficiência cardíaca. Dois tipos diferentes de amilóide são geralmente depositados no coração, dependendo da proteína antecedente defeituosa.
Amiloidose de AL: Esta desenvolve-se normalmente com base na discrasia de plasmócitos. As células plasmáticas clonais (por exemplo, no mieloma múltiplo) produzem um excesso de cadeias de luz livre. No caso de cadeias de luz amilóide, estas são depositadas como fibrilas amilóides em vários órgãos. O envolvimento cardíaco isolado é raro na amiloidose de AL. A maioria das vezes, os rins, o fígado e o sistema nervoso periférico são também afectados. As cadeias de luz Lambda são muito mais frequentemente detectadas como componentes amilóides do que as cadeias kappa.
Amiloidose ATTR: A transtiretina (TTR) é uma proteína de transporte, 98% da qual é produzida no fígado e é responsável pelo transporte de tiroxina e rethinol. O TTR é um tetrâmero que se pode dissociar em quatro monómeros. Normalmente, estes monómeros são solúveis no sangue. No entanto, se houver uma desordem, os monómeros agregam-se para formar fibrilhas amilóides patológicas. Existem duas causas de amiloidose TTR: A forma mutante (hereditária, familiar) ocorre devido a uma mutação no gene TTR. A prevalência das mutações difere de acordo com a região geográfica e grupos étnicos. A forma “selvagem” (adquirida, senil) geralmente só se manifesta numa idade mais avançada e afecta predominantemente o coração.
Manifestação clínica
Envolvimento cardíaco: Típico da amiloidose cardíaca é o desenvolvimento de insuficiência cardíaca com fração de ejeção preservada (HFpEF). Os depósitos amilóides tornam as paredes dos ventrículos e átrios mais espessas (sem hipertrofia real, uma vez que as células musculares do coração per se não são afectadas). Isto leva a um “endurecimento” do coração e a uma restrição do relaxamento (disfunção diastólica). Assim, há um aumento da pressão de enchimento diastólico final no ventrículo, dilatação dos átrios e um aumento da pressão pulmonar. Clinicamente, isto acaba por conduzir a insuficiência cardíaca esquerda e direita com dispneia, intolerância ao desempenho e edema. É também típico que o ventrículo rígido já não consiga ajustar correctamente o débito cardíaco. Devido ao volume do AVC ser fixado pela rigidez, o débito cardíaco só pode praticamente ser controlado pelo ritmo cardíaco. A síncope e a desregulamentação ortostática grave podem, portanto, ocorrer, especialmente sob stress, especialmente em pacientes que tomam beta-bloqueadores.
O átrio esquerdo alargado é um “bom” substrato para o desenvolvimento da fibrilação atrial. Pressões elevadas, juntamente com a baixa mobilidade do átrio devido a depósitos amilóides, podem levar a um aumento do risco de trombos intra-atriais e explicar o aumento significativo do risco de enfartes cerebrais em doentes com amiloidose.
Se os depósitos amilóides afectarem o sistema de condução, ocorrem bloqueios, especialmente bloqueios AV. Além disso, o risco de morte cardíaca súbita devido a arritmias ventriculares (especialmente fibrilação ventricular e “actividade eléctrica sem pulso” – PEA) é aumentado.
Outra manifestação típica da amiloidose cardíaca é a dor torácica sem que seja encontrada doença arterial coronária real. Por um lado, pequenas microtromboses na microcirculação podem desencadear angina pectoris (espasmos); por outro lado, a função endotelial perturbada revela uma disfunção microvascular.
Envolvimento de outros órgãos: O problema frequente da ortostatismo em doentes com amiloidose é geralmente causado por danos no sistema nervoso periférico e pela associada redução da vasoconstrição dos vasos periféricos.
A inervação intestinal perturbada pode levar a perturbações de passagem, flatulência, dor abdominal e irregularidades nas fezes. O envolvimento renal é geralmente manifestado pela proteinúria e por uma diminuição da função renal.
Os depósitos de amilóide no tracto gastrointestinal levam ocasionalmente à má absorção, perda de peso e aumento da rigidez do fígado. Na amiloidose AL, os sinais patognomónicos de hemorragia periorbital e macroglossia podem ocasionalmente (10%) ser encontrados. A amiloidose TTR é frequentemente precedida pela síndrome do túnel do carpo.
Diagnósticos
Se houver suspeita clínica de amiloidose cardíaca, são utilizados vários procedimentos de diagnóstico. O ECG mostra frequentemente baixa tensão periférica (atenuação da condução eléctrica aos eléctrodos devido à deposição amilóide), uma característica distintiva da cardiomiopatia hipertrófica, onde se observa normalmente um aumento da tensão. Imagens em bloco, fibrilação atrial e outras arritmias também podem ser vistas. No entanto, todas estas alterações não são específicas da doença. Para além dos parâmetros habituais, os biomarcadores cardiovasculares NT-proBNP e troponina devem ser medidos em laboratório. Um exame de urina, especialmente com a questão da albumina e da urina também não deve faltar. A electroforese de proteínas e a imunofixação no soro e na urina são também essenciais para distinguir as formas AL e ATTR.
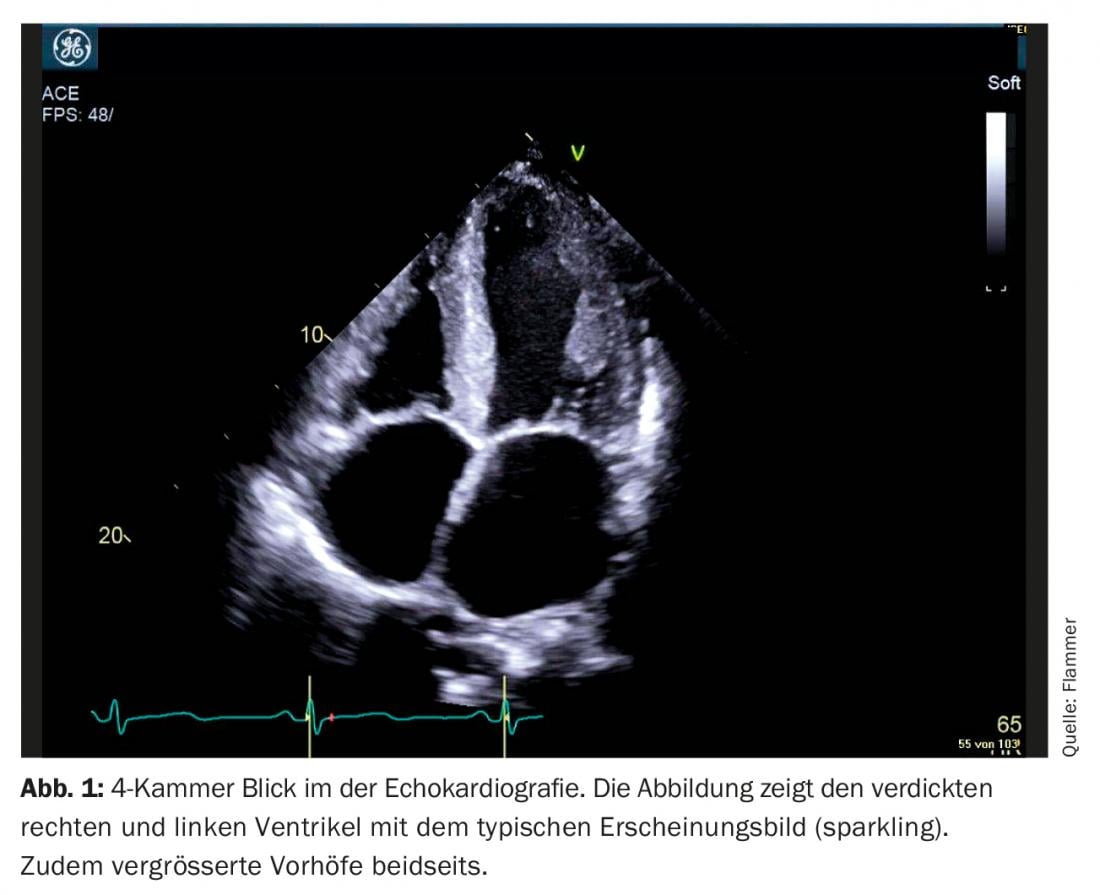
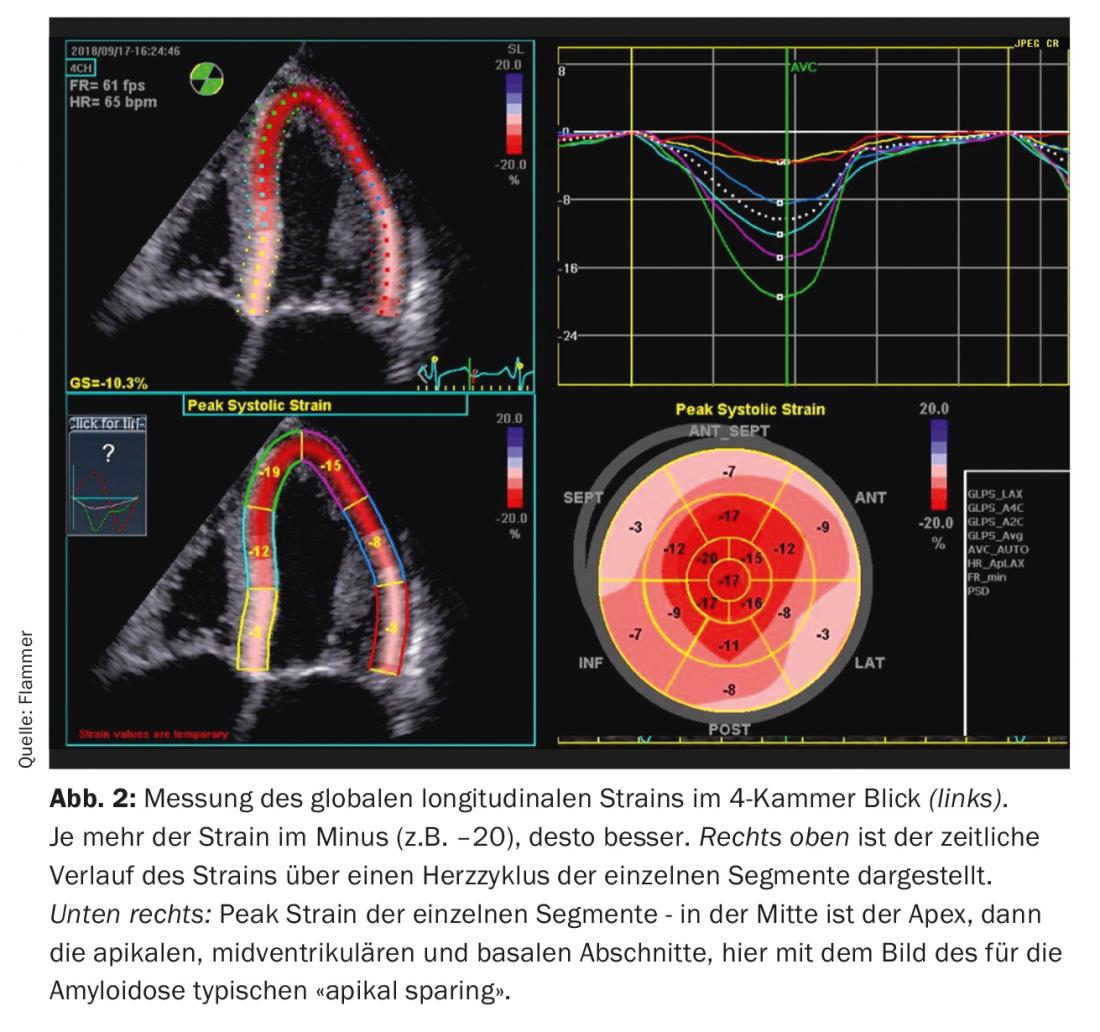
Ecocardiografia: O diagnóstico de amiloidose cardíaca é impensável sem imagem cardíaca. Os doentes com antecedentes e sinais de insuficiência cardíaca costumam receber ecocardiografia. Se houver sinais correspondentes de amiloidose no eco e insuficiência cardíaca manifesta, a amiloidose deve ser considerada. O que são sinais típicos na ecocardiografia? Há um miocárdio biventricularmente espessado, e o septo intra-atrial e as paredes atriais também são normalmente espessados. O miocárdio também se apresenta especificamente como “brilhante”, o que é característico da doença (contudo, é importante lembrar que a “segunda imagem harmónica” deve ser desligada na máquina de ultra-sons). O Amyloid também é depositado nas válvulas. Assim, parecem ligeiramente espessadas, mas a função das válvulas é normalmente normal. A efusão discreta do pericárdio também é comum (Fig. 1). Funcionalmente, especialmente em doenças avançadas, existe uma grave disfunção diastólica, geralmente com um padrão de enchimento restritivo. Devido ao aumento das pressões diastólicas associadas, ambos os átrios aumentam durante o curso da doença (dilatação biatrial) – característica de disfunção diastólica grave. Embora a fracção de ejecção seja normalmente normal, existe também uma disfunção sistólica, que se manifesta numa “deformação” reduzida (a deformação descreve a deformação medida do miocárdio). Em particular, a “tensão” longitudinal global é limitada. Típica e específica para a amiloidose é a chamada “poupa apical”, ou seja, a estirpe é normal (poupada) no ápice mas deteriora-se quanto mais se vai em direcção à base do coração (Fig. 2).
Ressonância magnética (MRI): Actualmente, a amiloidose é frequentemente diagnosticada utilizando este exame (por vezes como um achado incidental noutras investigações). As alterações observadas na RM são, em princípio, semelhantes às observadas no eco, mas podem ser observadas outras alterações típicas, especialmente se o gadolínio de contraste médio for também utilizado. Devido a uma lenta lavagem do gadolínio, este permanece mais tempo no interstício se o amilóide estiver presente. A ressonância magnética mostra então uma difusa e fragmentada “melhoria tardia do gadolínio” típica da amiloidose.
Algoritmo de diagnóstico para suspeita de amiloidose cardíaca
Em pacientes com um quadro clínico correspondente e com suspeita de amiloidose cardíaca, a discrasia de plasmócitos deve ser excluída antes de mais nada. Para tal, a electroforese de proteínas e a imunofixação devem ser procuradas na urina e no soro, tal como descrito acima. Se estiver presente, a pesquisa é principalmente para amiloidose AL, e na ausência de sinais, para amiloidose ATTR (Fig. 3).
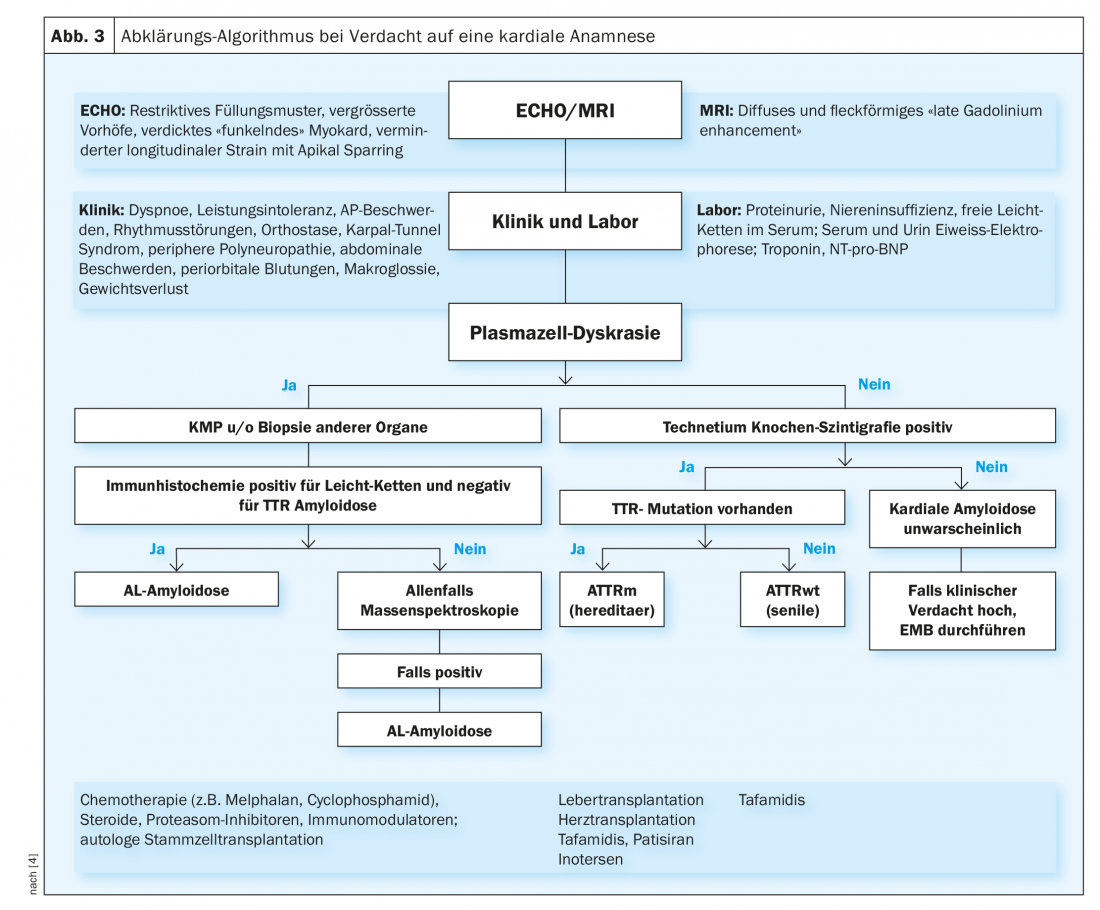
Se houver suspeita de amiloidose AL (discrasia de plasmócitos presente), deve ser feita uma tentativa de detectar a amiloidose AL directamente biotopicamente. Uma aspiração de medula óssea é geralmente realizada para diagnosticar a doença hematológica subjacente (geralmente mieloma múltiplo). Como a amiloidose é uma doença sistémica, o diagnóstico é confirmado assim que a amiloidose AL é detectada numa biópsia (de qualquer tecido). O menos invasivo e traumático é a aspiração de tecido adiposo abdominal. Além disso, o amilóide pode por vezes ser procurado em amostras de tecido previamente realizadas como parte de uma colonoscopia ou gastroscopia. Além disso, podem ser realizadas biópsias de glândulas salivares, lábios ou recto. Ocasionalmente, é também necessária uma biopsia renal ou endomiocárdica para se fazer um diagnóstico definitivo. Na amiloidose cardíaca, a biopsia endomiocárdica é muito sensível. Por um lado, as biópsias são examinadas para a amiloidose e, por outro, a imuno-histoquímica é utilizada para tentar classificar o tipo de amiloidose. Em casos difíceis, a espectroscopia de massa (padrão de ouro) é necessária. No entanto, apenas algumas patologias realizam um tal exame.
Se houver suspeita de amiloidose TTR (falta de provas de discrasia de plasmócitos), realiza-se hoje em dia uma cintilografia com tecnécio (osso). Nestas condições, a sensibilidade e especificidade desta metodologia na amiloidose ATTR cardíaca é muito boa. Se os resultados forem positivos, uma biópsia pode, portanto, ser omitida e o diagnóstico feito. Se a cintilografia não for clara e ainda houver suspeita de amiloidose cardíaca, deve ser feita uma biopsia endomilocárdica neste local.
Uma vez confirmada a amiloidose ATTR, são necessários testes genéticos para distinguir se se trata de um “tipo selvagem” ou de uma forma hereditária.
Terapia da amiloidose cardíaca
A insuficiência cardíaca está normalmente no centro dos sintomas da amiloidose cardíaca. Terapêuticamente, estes devem ser abordados antes de mais nada. Os diuréticos ajudam de forma sintomática. Especialmente porque as pressões de enchimento cardíaco são elevadas devido à fisiologia cardíaca restritiva, levando a uma elevação passiva da pressão pulmonar venosa. Os pacientes são significativamente menos sintomáticos sob diuréticos. No decurso da doença, são normalmente necessárias doses cada vez mais elevadas – em conformidade, é também importante assegurar que o potássio suficiente seja substituído. A titulação da dose é muitas vezes desafiante (caixa).

De acordo com a fisiologia HFpEF, não há indicação de inibidores da ECA, beta-bloqueadores e antagonistas de aldosterona. Pelo contrário, os inibidores da ECA, em particular, podem exacerbar o que já é muitas vezes pronunciado orthostasis. Os beta-bloqueadores resultam na incapacidade de aumentar o débito cardíaco sob stress, devido ao volume fixo do AVC. Assim, os beta-bloqueadores são retidos para pacientes em fibrilação atrial taquicárdica.
A fibrilação atrial e os enfartes cerebrais como resultado são comuns. Por um lado, é essencial não perder o VHF (exames Holter semestrais), por outro lado, a anticoagulação oral deve ser iniciada se estiver presente, independentemente da pontuação CHADS-Vasc. Não é completamente claro se o OAK deve ser iniciado na ausência do FCR – isto é provavelmente apropriado no caso de um padrão de enchimento restritivo. Em VHF taquicárdico, o controlo da taxa com beta-bloqueador deve ser feito, a digoxina não deve ser administrada devido ao aumento da toxicidade. A amiodarona é indicada em certos pacientes para o controlo do ritmo e ocasionalmente para o controlo da taxa.
Devido às perturbações no sistema de condução cardíaca, torna-se necessário o implante de pacemaker, especialmente em pacientes com amiloidose TTR, uma indicação que pode ser dada generosamente. A questão de implantar ou não um cardioversor desfibrilador implantável (CDI) é particularmente difícil. A causa mais comum de morte cardíaca em pacientes com amiloidose é morte cardíaca súbita devido a arritmias ventriculares. Não só se observa fibrilação ventricular, mas também um número relevante de EPAs. Estes últimos não são detectados de forma fiável por um CDI. Há também uma taxa mais elevada de choques aplicados incorrectamente (o que é muito traumático para os pacientes) e de partos sem êxito de choques. Até agora, existem apenas estudos retrospectivos sobre este tópico, que não mostram nenhum benefício claro para um CDI. A indicação deve, portanto, ser feita com cautela e discutida em pormenor numa equipa de amiloidose.
Terapia causal
Na amiloidose AL, a doença hematológica deve ser abordada em primeiro lugar e acima de tudo. Contudo, isto é frequentemente desafiante, especialmente em casos de envolvimento cardíaco grave e de insuficiência cardíaca grave concomitante. A terapia de alta dose com transplante autólogo de células estaminais, que muitas vezes se destina a transplante, não pode ser realizada neste caso. Os outros regimes terapêuticos com agentes quimioterápicos (por exemplo melphalan, ciclofosfamida), esteróides e novos inibidores do proteasoma, imunomoduladores ou anticorpos anti-CD38 também podem ocasionalmente ser stressantes para o coração. No entanto, há abordagens curativas em alguns casos. No entanto, um tratamento detalhado iria para além do âmbito deste artigo.
Para a amiloidose TTR hereditária, o transplante hepático (e o transplante cardíaco em fases avançadas) continua a ser o tratamento de escolha.
Contudo, desde o Verão de 2018, dois medicamentos estão disponíveis nos EUA e na Europa (aprovação pela FDA e EMA) que utilizam a tecnologia de interferência do ARN para “silenciar” o gene defeituoso de modo a que não seja produzido TTR amiloidogénico. Nenhum dos dois medicamentos foi ainda aprovado na Suíça. Um estudo em grande escala em doentes com amiloidose hereditária mostrou de forma impressionante como a polineuropatia (que é frequentemente prevalecente na amiloidose hereditária) pode ser travada. As análises de subgrupos do partisiran de drogas mostram que os resultados no coração poderiam ser igualmente bons, mas ainda estão por ver mais estudos. Até há pouco tempo, não havia terapia causal para amiloidose TTR do tipo selvagem. Felizmente, um estudo também publicado no Verão de 2018 mostrou que o tafamidis, um estabilizador de transthyretin, que também é aprovado na Europa e nos EUA para o tratamento da neurpatia amilóide hereditária, tem bons resultados na amiloidose cardíaca com melhoria da qualidade de vida, redução da mortalidade e diminuição das hospitalizações relacionadas com o coração. O medicamento ainda não está actualmente disponível na Suíça, mas espera-se a sua aprovação em breve devido aos bons resultados.
Mensagens Take-Home
- A amiloidose AL é uma doença rara que é normalmente desencadeada por uma doença hematológica subjacente de “cadeia leve” e que muitas vezes afecta outros órgãos para além do coração. No centro da terapia está o tratamento da doença hematológica.
- A amiloidose TTR pode ser causada por uma mutação no gene TTR ou pode ser “adquirida” no decurso da vida (“tipo selvagem”). A forma familiar mais rara manifesta-se predominantemente com polineuropatia ou cardiopatia (ou uma combinação), dependendo do gene envolvido; a forma “tipo selvagem” está normalmente confinada ao coração. Além da terapia sintomática, existem novas abordagens terapêuticas promissoras com estabilizadores TTR e moléculas de interferência de RNA.
- O diagnóstico de amiloidose cardíaca requer uma clínica apropriada (insuficiência cardíaca), eco ou resultados de ressonância magnética típicos de amiloidose e ou evidência de amiloide numa biopsia ou uma cintilografia Tc positiva.
- O quadro clínico típico da amiloidose cardíaca inclui, em particular, sintomas de insuficiência cardíaca, ortoestase, síncope, arritmias (especialmente VHF e taquicardia ventricular/fibrilação ventricular), bloqueio AV e angina microvascular.
Literatura:
- Brouwers S, et al: Cardiac amyloidosis Cardiovasc Med. 2018; 21(11): 282-289.
- Rauch PJ, et al: Systemic amyloidoses Switzerland Med Forum 2014 14; 943-948.
- Laptseva N, et al: Amiloidose cardíaca: ainda a desafiar o Eur Heart J 2017, 38(22): 122.
- Falk RH, et al: AL (Light-Chain) Cardiac Amiloidosis: A Review of Diagnosis and Therapy.
- Gillmore JD, et al: Nonbiopsy Diagnosis of Cardiac Transthyretin Amyloidosis Circulation 2016: j133(24): 2404-2412.
- Maurer MS, et al: Tafamidis Tratment for Patients with Transthyretin Amyloid Cardiomyopathy N Engl J Med 2018; 379(11): 1007-1016.
- Adams D, et al: Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis N Engl J Med 2018; 379(1): 11-21.
CARDIOVASC 2019; 18(2): 6-10