A doença pulmonar intersticial é relativamente rara na prática geral. No entanto, deve ser considerada em casos de dispneia crónica, tosse seca e balanços finos de bolhas. O diagnóstico precoce e correcto é crucial.
As doenças pulmonares intersticiais (DPI) subsumem um grande grupo de diferentes doenças raras do parênquima pulmonar com apresentação clínica, radiológica e patológica semelhante [1]. Os processos subjacentes ao ILD, o prognóstico, mas também as terapias diferem consideravelmente de entidade para entidade. Por um lado, encontramos doenças com um prognóstico favorável, mas também doenças com um curso agudo e elevada mortalidade. A fibrose pulmonar idiopática (IPF), a DPI mais comum entre as pneumonias intersticiais idiopáticas (PII), mostra um curso comparável aos tumores malignos agressivos com uma sobrevivência mediana de cerca de três anos após o diagnóstico [2].
O tratamento da DPI em pacientes individuais depende em grande parte da entidade da doença. Para alguns padrões de doença, o foco é a abstinência de exposição a substâncias nocivas (inalatórias). Dependendo da entidade, são utilizadas terapias imunossupressoras ou, no caso da IPF, terapia antifibrótica. Um diagnóstico precoce e correcto, com especial reconhecimento dos factores reversíveis, é crucial para um óptimo aconselhamento e tratamento dos pacientes.
Classificação das doenças pulmonares intersticiais
A DPI pode ser dividida em doenças de causa conhecida e doenças de causa desconhecida (pneumonia intersticial idiopática, IIP) (Fig. 1) . A lista de possíveis causas de DPI é longa. Entre as causas conhecidas, encontramos exposições a poeiras inorgânicas ou orgânicas, medicamentos e raios X. A complicação da DPI pode ocorrer em muitas doenças reumatológicas. A DPI pode ser a primeira manifestação de uma doença reumatológica subjacente antes de poder ser diagnosticada utilizando os critérios habituais de diagnóstico. Em 2015, o termo “pneumonia intersticial com características auto-imunes” (IPAF) foi introduzido para reflectir esta situação [3].
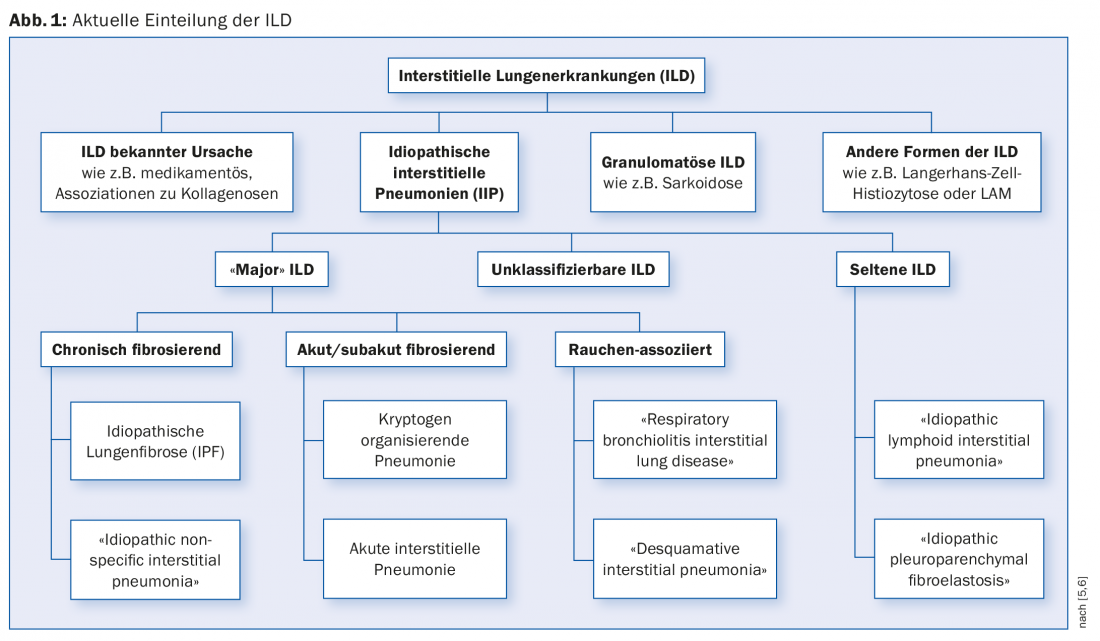
A causa da PII é – como o nome sugere – pouco clara. As PII “principais” são classificadas de acordo com o curso clínico em fibrose crónica, fibrose aguda/subaguda e doenças associadas ao tabagismo.
A designação “idiopático” é parcialmente contraditória com a fisiopatologia no grupo das doenças associadas ao fumo, pelo que se discute uma categorização fora do PII. O fumo provoca alterações histologicamente detectáveis (“bronquiolite respiratória”). Os indivíduos suspeitos podem desenvolver DPI (“bronquiolite respiratória intersticial pulmonar” [RB-ILD] e “desquamative interstitial pneumonia”) [4].
Epidemiologia do ILD
As DPI são doenças raras, pelo que há poucos dados epidemiológicos. Um grande estudo epidemiológico em 1994 encontrou uma incidência de 31,5 casos por 100.000/ano em homens, e 26,1 casos por 100.000/ano em mulheres [7]. O IPF, que é mais comum nos homens mais velhos, é a entidade mais comum entre as IILDs. Os dados epidemiológicos para o IPF na Suíça não existem. Estima-se que existam 100 a 5000 doentes com IPF na Suíça (prevalência de 1,25-63 casos/100.000) [8]. A idade no primeiro diagnóstico de DPI varia entre as diferentes entidades da doença. Enquanto a incidência de IPF aumenta significativamente com a idade, as sarcoidoses, as histiocitoses celulares de Langerhans ou, nas mulheres, as linfangioleiomiomatoses (LAM) são mais comuns na vida adulta jovem.
Diagnóstico de ILD
O ILD é diagnosticado tendo em conta os resultados clínicos, radiológicos, pulmonares, químicos laboratoriais e, dependendo da situação, citológicos/histológicos. O diagnóstico e a determinação da estratégia terapêutica para o paciente individual são realizados de forma óptima num quadro interdisciplinar composto por clínicos (pneumologistas), radiologistas e patologistas (conselho ILD). Está provado que se pode conseguir uma melhoria qualitativa significativa no diagnóstico [9].
Clínica: A clínica de ILD é frequentemente inespecífica, com uma dispneia que aumenta lentamente e geralmente uma tosse seca. O exame clínico revela muitas vezes escalas de bolhas finas e basicamente acentuadas e, particularmente em doenças avançadas, saturação reduzida de oxigénio (especialmente ao esforço) e sinais de hipoxemia crónica, tais como unhas de vidro de relógio e dedos de coxinha. É importante considerar a presença de DPI cedo no diagnóstico diferencial de pacientes com estes sintomas.
Muitas vezes, uma história cuidadosa fornece-nos pistas decisivas sobre a entidade da DDI. A duração dos sintomas, mas também as possíveis influências ambientais, incluindo um histórico profissional detalhado, são importantes. A identificação de influências ambientais potencialmente reversíveis é de grande importância. O reconhecimento de possíveis aeroalergénios que podem desencadear uma pneumonite de hipersensibilidade (alveolite alérgica exógena, EAA) é elementar tanto para o diagnóstico como para a eliminação da exposição decisiva. Na maioria das vezes encontramos a criação de aves, contacto com a agricultura ou exposição a bolores. Fumar é um desencadeador potencialmente reversível na DPI associada ao fumo. Para além das doenças pleurais associadas ao amianto, a exposição ao amianto também pode levar à fibrose pulmonar associada ao amianto. Os sintomas de uma doença reumatológica subjacente devem ser especificamente procurados. Muitos medicamentos podem causar ILD como um efeito secundário. Neste ponto, gostaríamos de consultar a página inicial www.pneumotox.com, que é muito útil na vida quotidiana e fornece informações sobre os efeitos secundários tóxicos pulmonares conhecidos.
O histórico familiar deve ser tomado; em até 20% de todos os casos de ILD há uma forma familiar com as mutações genéticas correspondentes [10].
Imagiologia: A radiografia convencional do tórax sugere frequentemente doença pulmonar intersticial com aumento das marcações reticulares e/ou nodulares do pulmão.
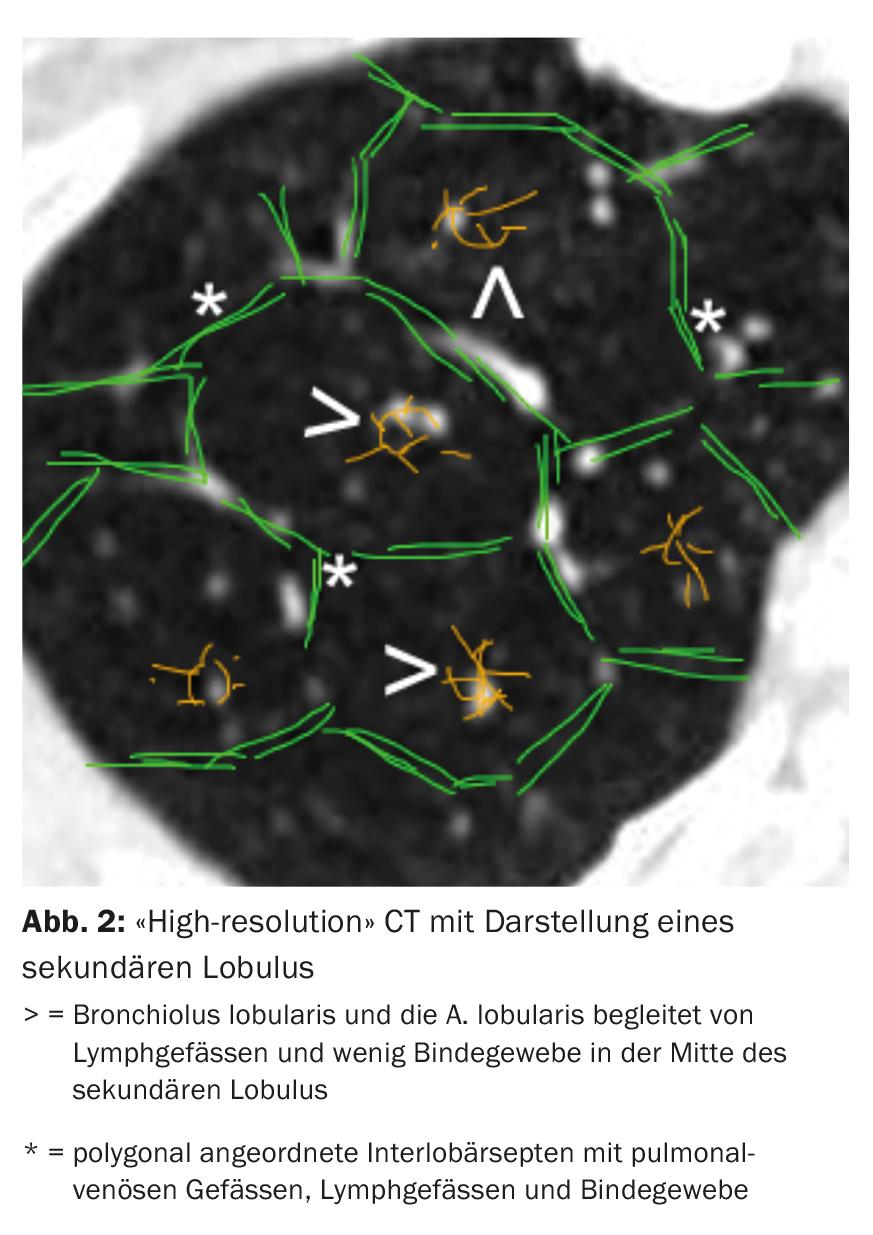
A tomografia computorizada (CT) do tórax com cortes de “alta resolução” desempenha um papel central no diagnóstico da DPI. Com base no tipo e disposição das alterações em relação à localização do lóbulo secundário (Fig. 2) e à topografia pulmonar, podem distinguir-se diferentes padrões radiológicos.
O padrão radiológico único não é específico de uma entidade de ILD. Por exemplo, no IPF encontramos um padrão “habitual de pneumonia intersticial” (PIU) com reticulações subpleurais basais acentuadas, padrão de favo de mel acentuado basal e bronquiectasia de tracção (Fig. 3) [11]. No entanto, um padrão UIP também pode ser encontrado noutras entidades, tais como a vasculite de pequenos vasos, CEA crónica, ILD no cenário de artrite reumatóide ou asbestose.
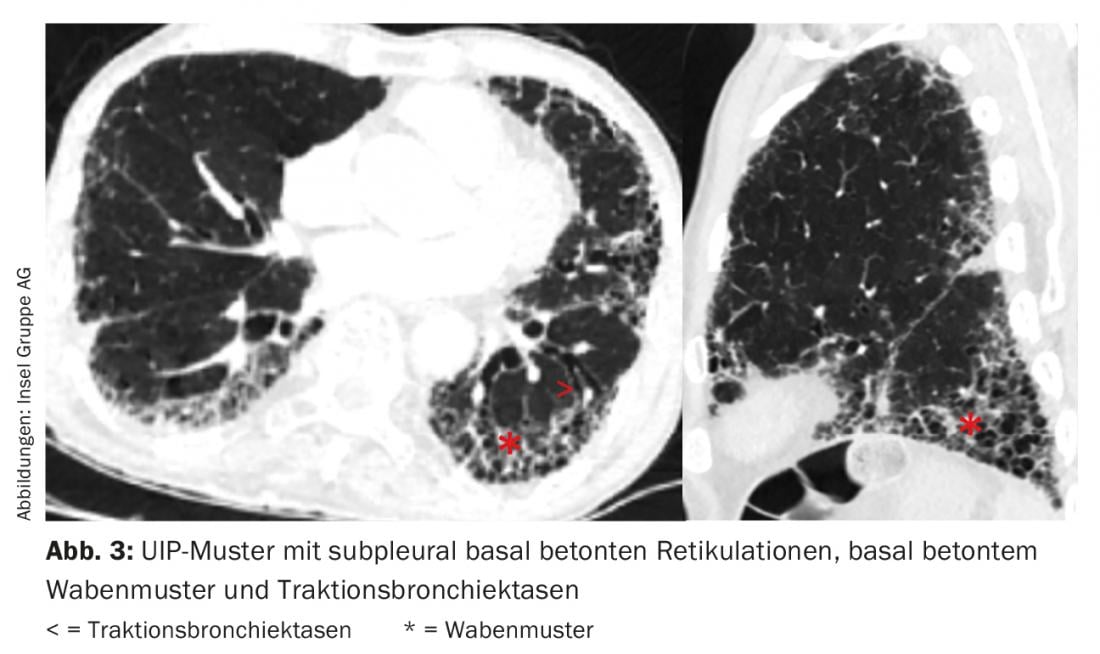
Função pulmonar: Em ILD encontramos normalmente restrição e troca de gás prejudicada (capacidade de difusão). À medida que a doença progride, existe inicialmente hipoxemia induzida pelo exercício e, à medida que a doença progride, hipoxemia induzida pelo repouso.
A capacidade vital forçada (CVF) é utilizada como um parâmetro importante de progressão. Uma queda de CVF de ≥10% em 24 semanas aumenta o risco de mortalidade para os próximos doze meses num factor de 4,8 [12]. Para além do CVF facilmente medido, a capacidade de difusão, o teste de caminhada de 6 minutos e os índices multivariados correlacionam-se também com o prognóstico [13].
Laboratório: O laboratório pode ser útil na classificação de ILD. Para além de um hemograma (eosinofilia), a função renal, os valores hepáticos e inflamatórios, bem como a pesquisa serológica de uma doença reumatológica subjacente, podem dar-nos indicações.
Investigações invasivas: Como regra, uma broncoscopia com pelo menos um lavado broncoalveolar (BAL) é realizada em pacientes com um novo diagnóstico de DPI. Um BAL cada vez mais hemorrágico de parte a parte é típico da hemorragia alveolar (por exemplo, na vasculite de pequenos vasos). A distribuição celular no BAL mostra alterações características em algumas entidades ILD (por exemplo eosinofilia na pneumonia eosinofílica, linfocitose na sarcoidose e EAA). Dependendo do diagnóstico suspeito baseado na apresentação clínica e radiológica, são realizadas biópsias de gânglios linfáticos mediastinais/hilares e/ou biópsias de pulmões transbrônquicos. Os fragmentos de tecido das biópsias pulmonares transbrônquicas são frequentemente demasiado pequenos para um diagnóstico fiável. Amostras maiores de tecido podem ser obtidas com toracoscopia ou, desde há alguns anos, também broncoscopicamente com criobiopsia.
Terapia
A terapia de ILD difere fundamentalmente de entidade para entidade (Fig. 4) . Em algumas entidades, por exemplo, a doença pode ser decisivamente influenciada pela abstinência de exposição (cessação do tabagismo, abstinência de desencadeadores de alveolite alérgica exógena, mudança de medicação). Muitas DPI são tratadas com imunossupressão, tais como doenças associadas a colagenoses/vasculites, (criptogénicas) pneumonias organizadoras, pneumonias eosinofílicas ou sarcoidoses. A imunossupressão é geralmente feita com corticosteróides sistémicos, muitas vezes em combinação com um fármaco com esteróides (por exemplo, azatioprina ou micofenolato).
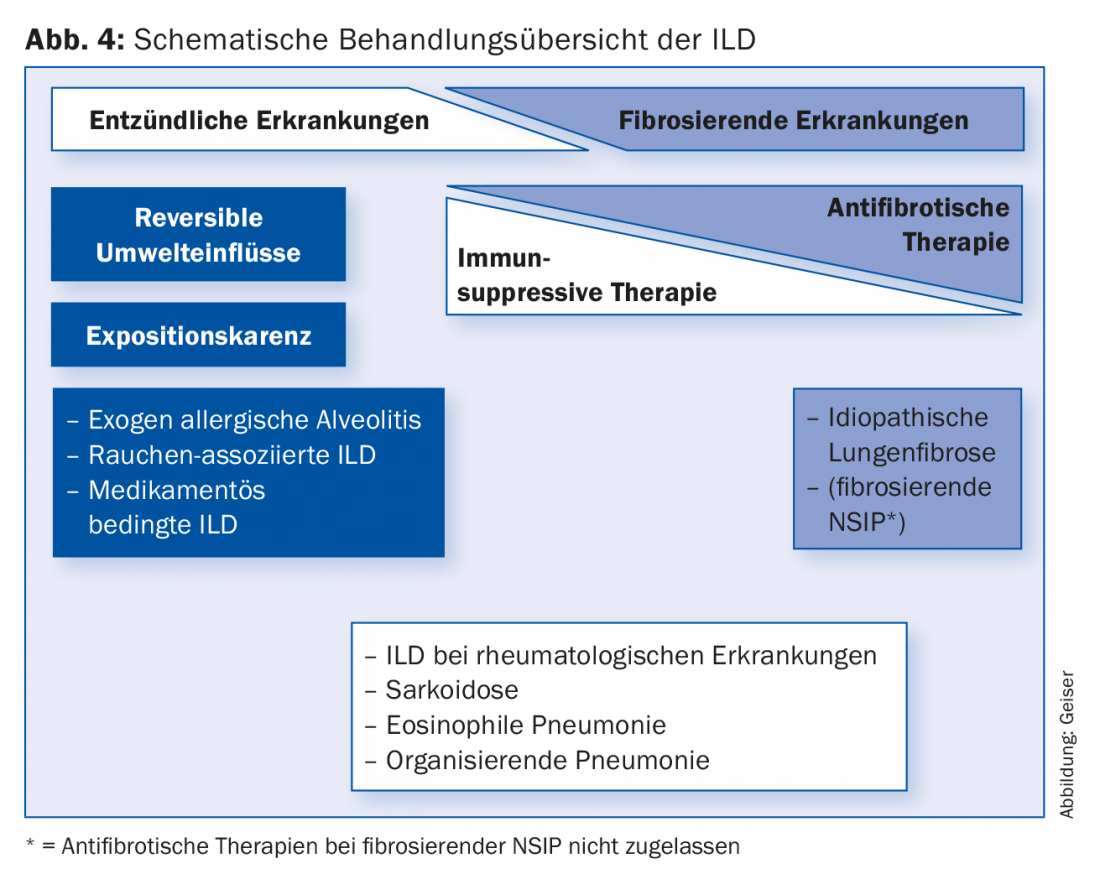
As terapias imunossupressoras comummente utilizadas para o IPF até há alguns anos atrás já não são recomendadas [14,15]. Após muitos estudos negativos com várias substâncias, os dois medicamentos actualmente aprovados para o tratamento da IPF, pirfenidona (Esbriet®) e nintedanibe (Ofev®), demonstraram retardar a progressão da doença [16–18]. Novos dados sugerem que a sobrevivência pode ser favoravelmente influenciada.
Em pacientes mais jovens com entidades ILD em rápido progresso, tais como a IPF, a avaliação para transplante pulmonar deve ocorrer cedo no curso da doença. À medida que a DPI progride, a oxigenoterapia a longo prazo para doentes hipoxémicos e outras terapias paliativas, incluindo a reabilitação pulmonar, pode ser necessária.
Mensagens Take-Home
- Embora a doença pulmonar intersticial (DPI) seja relativamente incomum no contexto da prática geral, a dispneia crónica, a dispneia seca
- A tosse e as balanças de bolhas finas devem ser consideradas como um diagnóstico diferencial.
- Entre as ILDs, encontramos entidades de doenças muito diferentes com cursos diferentes.
- Um diagnóstico precoce e correcto é crucial para um óptimo aconselhamento e tratamento do doente.
- A terapia de DPI difere significativamente dependendo da entidade da doença e varia desde a abstinência de exposição aos agentes desencadeantes até à imunossupressão e ao uso de terapias antifibróticas na fibrose pulmonar idiopática (IPF).
- Os pacientes mais jovens com uma entidade ILD em rápido progresso devem ser avaliados para transplante pulmonar numa fase inicial.
- No curso, a oxigenoterapia a longo prazo em doentes hipoxémicos e outras terapias paliativas podem tornar-se necessárias.
Literatura:
- Cosgrove G, Schwarz M: Abordagem à Avaliação e Diagnóstico da Doença Pulmonar Intersticial. In: Schwarz M, King T (eds.): Interstitial Lung Disease. 5ª ed. Shelton: People’s Medical Publishing House-USA 2011: 3-34.
- Latsi PI, et al: Fibrotic idiopathic interstitial pneumonia: o valor prognóstico das tendências funcionais longitudinais. Am J Respir Crit Care Med 2003; 168(5): 531-537.
- Fischer A, et al: Uma declaração oficial de investigação da Sociedade Respiratória Europeia/Sociedade Torácica Americana: pneumonia intersticial com características auto-imunes. Eur Respir J 2015; 46(4): 976-987.
- Fraig M, et al: Bronquiolite respiratória: um estudo clinicopatológico em fumadores actuais, ex-fumadores, e nunca-fumadores. Am J Surg Pathol 2002; 26(5): 647-653.
- American Thoracic Society/European Respiratory Society: International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias. Am J Respir Crit Care Med 2002; 165(2): 277-304.
- Travis WD, et al: Uma declaração oficial da American Thoracic Society/European Respiratory Society: Actualização da classificação internacional multidisciplinar das pneumonias intersticiais idiopáticas. Am J Respir Crit Care Med 2013; 188(6): 733-748.
- Coultas DB, et al: A epidemiologia das doenças pulmonares intersticiais. Am J Respir Crit Care Med 1994; 150(4): 967-972.
- Funke M, Geiser T: Fibrose pulmonar idiopática: o ponto de viragem é agora! Swiss Med Wkly 2015; 145: w14139.
- Flaherty KR, et al: Pneumonia intersticial idiopática: qual é o efeito de uma abordagem multidisciplinar do diagnóstico? Am J Respir Crit Care Med 2004; 170(8): 904-910.
- Kropski JA, Blackwell TS, Loyd JE: A base genética da fibrose pulmonar idiopática. Eur Respir J 2015; 45(6): 1717-1727.
- Raghu G, et al: Uma declaração oficial ATS/ERS/JRS/ALAT: fibrose pulmonar idiopática: orientações baseadas em provas para o diagnóstico e gestão. Am J Respir Crit Care Med 2011; 183(6): 788-824.
- du Bois RM, et al: Capacidade vital forçada em pacientes com fibrose pulmonar idiopática: propriedades do teste e diferença clinicamente mínima importante. Am J Respir Crit Care Med 2011; 184(12): 1382-1389.
- Sharp C, Adamali HI, Millar AB: Uma comparação de índices multidimensionais publicados para prever o resultado em fibrose pulmonar idiopática. ERJ Open Res 2017; 3(1). DOI: 10.1183/23120541.00096-2016.
- Funke M, et al: Idiopathic Pulmonary Fibrosis in Switzerland: Diagnosis and Treatment (Fibrose Pulmonar Idiopática na Suíça: Diagnóstico e Tratamento). Respiração 2017; 93(5): 363-378.
- Raghu G, et al: Uma Directriz Oficial de Prática Clínica ATS/ERS/JRS/ALAT: Tratamento da Fibrose Pulmonar Idiopática. Uma actualização da Directriz da Prática Clínica de 2011. Am J Respir Crit Care Med 2015; 192(2): e3-19.
- Noble PW, et al: Pirfenidone em doentes com fibrose pulmonar idiopática (CAPACIDADE): dois ensaios aleatórios. Lancet 2011; 377(9779): 1760-1769.
- Richeldi L, et al: Eficácia e segurança do nintedanib na fibrose pulmonar idiopática. N Engl J Med 2014; 370(22): 2071-2082.
- King TE Jr, et al: Um ensaio fase 3 de pirfenidona em doentes com fibrose pulmonar idiopática. N Engl J Med 2014; 370(22): 2083-2092.
PRÁTICA DO GP 2017; 12(12): 13-18