A hepatite auto-imune (AIH) é uma doença hepática crónica e progressiva caracterizada pelo quadro histológico da hepatite de interface, hipergamaglobulinaemia e auto-anticorpos circulantes. A AIH ocorre em todos os grupos etários e afecta mais frequentemente as mulheres do que os homens (proporção 3:1). O espectro da AIH varia de doença assintomática a hepatite aguda ou mesmo fulminante, a insuficiência hepática. Os corticosteróides são utilizados para induzir a remissão. Na terapia de manutenção, a azatioprina é preferível para reduzir o risco de recorrência durante pelo menos dois, de preferência quatro anos.
A hepatite auto-imune (AIH) é responsável por 11-23% de todas as hepatopatias crónicas. A incidência pode ser subestimada porque a hepatite viral é comum e quando a hepatite viral crónica está presente, qualquer hepatite autoimune concomitante pode passar despercebida. A incidência anual em caucasianos é de 0,1-1,9/100 000, e a prevalência é de 16,9/100 000 [1]. A AIH é responsável por 2,6% dos transplantes de fígado na Europa [2] e por 4-6% dos transplantes de fígado nos EUA [3]. As mulheres são afectadas cerca de três vezes mais frequentemente. Basicamente, a doença ocorre em todos os grupos etários, mas em 50% de todos os casos, a doença começa antes dos 30 anos de idade.
Patogénese
A patogénese exacta da AIH continua a não ser clara. O quadro clínico reflecte uma interacção complexa entre predisposição genética, factores desencadeantes, auto-antigénios e mecanismos imunoregulatórios, conduzindo em última análise a um processo imunológico (mediado por células, mediado por anticorpos ou em combinação) contra hepatócitos com desenvolvimento de inflamação crónica que conduz à cirrose. Os factores de desencadeamento exactos são desconhecidos; agentes infecciosos, medicinais e tóxicos são possíveis. A mímica molecular de antigénios estranhos e autogénios é a explicação mais comum para a perda de auto-tolerância [4].
Clínica
A AIH tem uma aparência muito heterogénea e flutuante. O diagnóstico é frequentemente atrasado porque muitas vezes apenas estão presentes sintomas ligeiros, não específicos de hepatite aguda e auto-limitada. O Quadro 1 dá uma visão geral das queixas típicas e dos resultados clínicos [5].
Existem diferentes formas de manifestação, que são brevemente discutidas a seguir:
- Assintomático: O diagnóstico é feito com base num achado incidental de enzimas hepáticas elevadas em doentes assintomáticos.
- Hepatite aguda: Início agudo em cerca de 40% dos casos. Uma anamnese detalhada revela frequentemente sintomas anteriores não específicos tais como episódios de mal-estar, náuseas e artralgia.
- Insuficiência hepática aguda: Em casos raros, apresentação inicial como hepatite fulminante (5%) ou descompensação hepática (“doença queimada”) com ascite (20%), encefalopatia hepática (14%) e varizes hepáticas hemorrágicas de esófago (8%).
- Cirrose hepática: Até 30% dos diagnósticos iniciais como cirrose hepática ou com complicações correspondentes.Uma característica típica da AIH é a sua associação com síndromes imunomédicas extra-hepáticas como a tiroidite auto-imune, vitiligo, alopecia, colite ulcerosa, artrite reumatóide, diabetes mellitus e glomerulonefrite.
Divisão
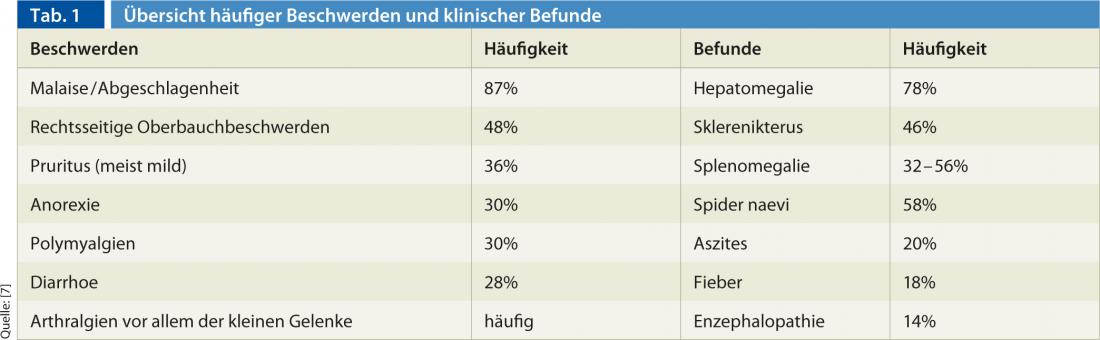
Com base na constelação de anticorpos, a AIH é classificada em três tipos diferentes (tipo I-III) (Quadro 2). A AIH tipo I é a AIH “clássica” e a forma mais comum em todo o mundo. Em comparação, a AIH tipo II é rara e afecta principalmente doentes pediátricos (raparigas/jovens). A ocorrência de AIH pode ocorrer em combinação com outras doenças auto-imunes do fígado (as chamadas “síndromes de sobreposição”). A AIH sobrepõe-se mais frequentemente à cirrose biliar primária positiva da AMA (cerca de 88%), mas a colangite esclerosante primária e a hepatite viral também podem estar presentes ao mesmo tempo.
Diagnósticos
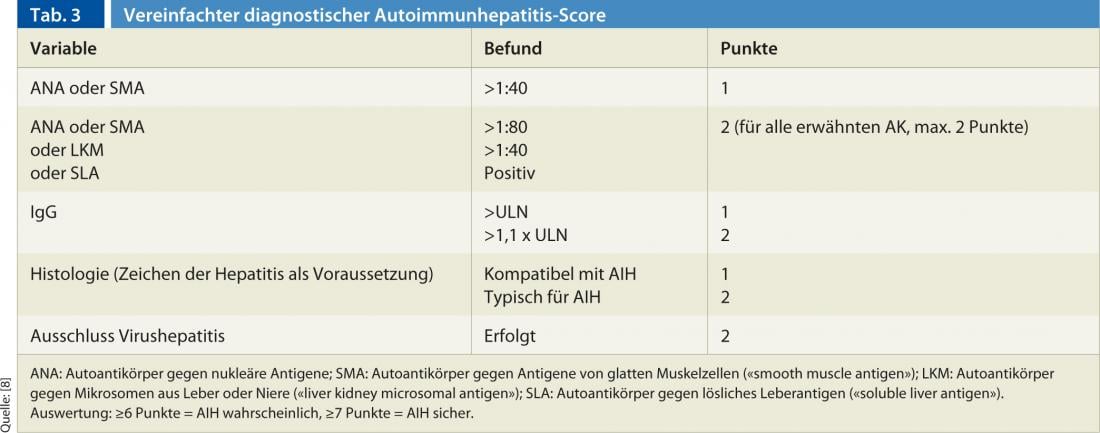
O diagnóstico baseia-se numa combinação de achados clínicos, histológicos e serológicos, bem como na exclusão de outras hepatopatias crónicas (principalmente hepatite viral, hepatite induzida por drogas, hepatopatias hereditárias e metabólicas) [7]. Nenhuma descoberta única é patognomónica para a AIH. Por esta razão, várias pontuações foram estabelecidas no passado, com a pontuação recentemente publicada e simplificada por Hennes et al. é fácil de manusear na prática clínica diária [8]. A pontuação distingue entre um diagnóstico definitivo (mín. 7 pontos) e um provável (mín. 6 pontos) de AIH e baseia-se nos quatro critérios seguintes (Quadro 3):
- Presença de auto-anticorpos
- Imunoglobulina G (IgG) elevada
- Histologia do fígado
- Exclusão da hepatite viral
A sensibilidade e especificidade desta pontuação são elevadas: para um diagnóstico definitivo, os valores são 81% e 99%, respectivamente; para um diagnóstico provável, 88% e 97% [8].
Laboratório: Os exames químicos de laboratório mostram geralmente o quadro típico de danos hepatocelulares do fígado: as transaminases são claramente superiores aos parâmetros de colestase (AST: rácio ALP >3). Um quadro colestático com hiperbilirrubinemia directa predominante e elevação de ALP é mais raro. Além disso, em caso de suspeita de AIH, devem ser determinadas as globulinas gama e os auto-anticorpos contra antigénios nucleares (ANA), contra antigénios de células musculares lisas (“antigénio do músculo liso” [SMA]) e contra microssomas do fígado ou rim (“antigénio do microssoma do rim do fígado” [LKM]).
Autoanticorpos adicionais mais recentes incluem anticorpos contra o citosol do fígado 1 (LC-1), cujos títulos se correlacionam com a actividade inflamatória da AIH, ou anticorpos receptores asialoglicoproteicos (ASGPR). Estes podem ser detectados em até 90% do tipo I de AIH e têm significado prognóstico.
Histologia: A histologia é essencial para fazer um diagnóstico definitivo, mesmo que os achados histológicos não sejam específicos para a AIH [5]. Mesmo que a apresentação clínica seja aguda, histologicamente já existem normalmente sinais de hepatopatia crónica. A expressão histológica não está correlacionada com o grau de elevação dos valores hepáticos ou IgG. A histologia é assim um parâmetro mais fiável do que os resultados laboratoriais para estimar a gravidade da doença [9].
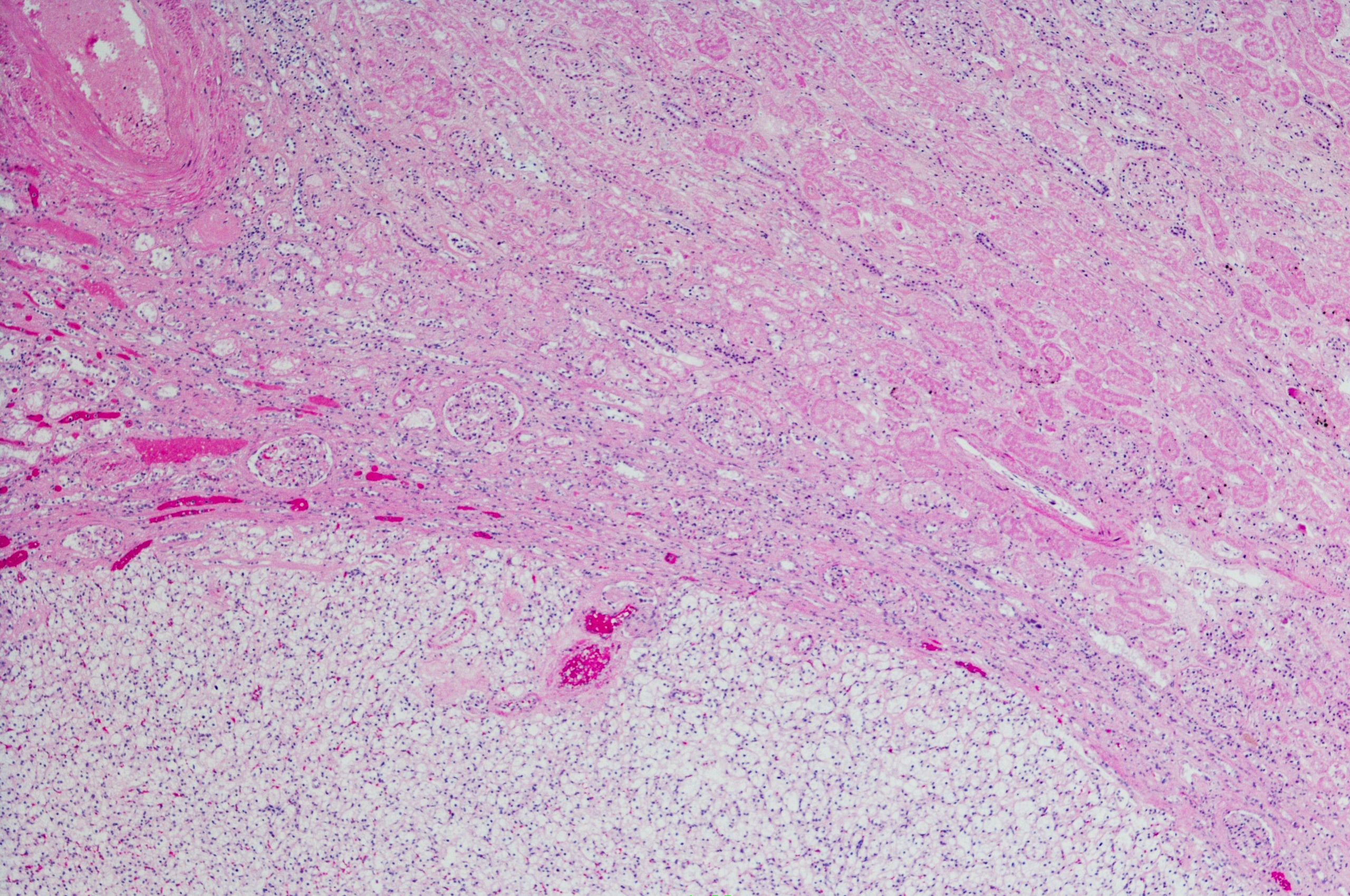
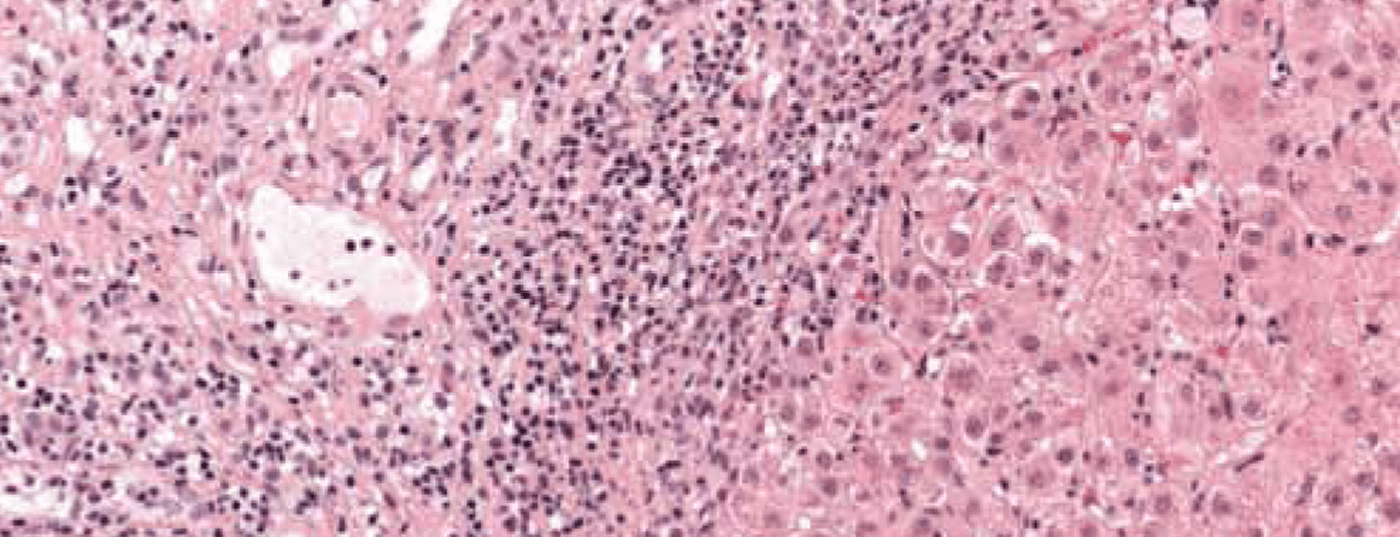
A imagem de uma doença necroinflamatória crónica é característica: portal e periportal (“necrose fragmentada”, “hepatite de interface”) bem como infiltrados monocíticos (linfoplasmocíticos) (Fig. 1). Em doenças graves e avançadas, existe uma extensa necrose e fibrose de ponte. A “hepatite de interface” não indica necessariamente um curso progressivo da doença, enquanto que a “necrose de ponte” aumenta a probabilidade de progressão para a cirrose. A especificidade do achado histológico global é de 81%, o valor preditivo positivo é de 68%. A diferenciação da hepatite induzida por drogas ou viral da AIH pode, portanto, por vezes ser difícil.
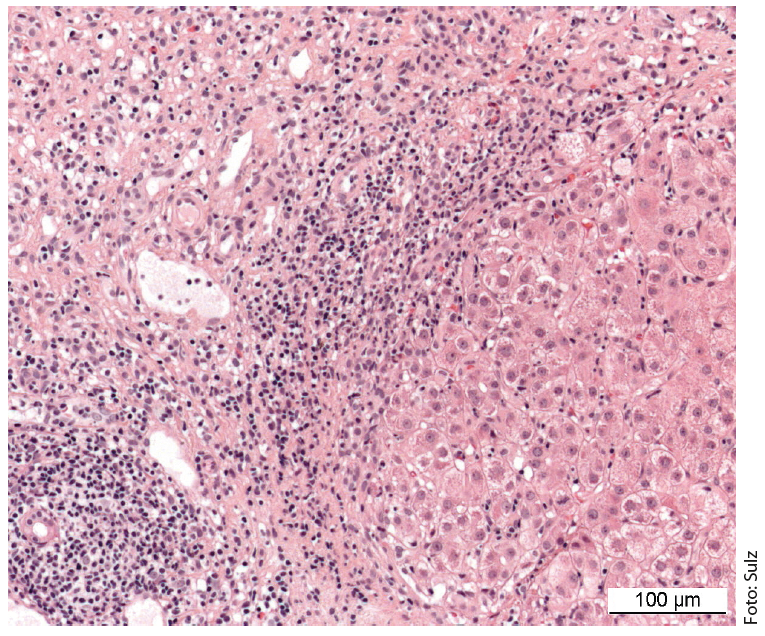
Fig. 1: A histologia hepática de uma menina de 12 anos mostra secções parcialmente fibrosadas do campo portal com infiltrados inflamatórios linfoplásicos densos e hepatite de interface activa. Coloração: hematoxilina-eosina.
Terapia
Indicações: De acordo com a Associação Americana para o Estudo das Doenças Hepáticas (AASLD), existem indicações absolutas e relativas para o tratamento da AIH (Quadro 4) [3].

Classificação da terapia: A terapia pode ser dividida em diferentes fases:
- Indução de remissão
- Manutenção de remissão
- Afunilamento ou descontinuação
- Terapia de uma recaída
Objectivo terapêutico: O objectivo da terapia é a remissão permanente. Classicamente, a remissão é definida como uma queda na AST <2 vezes a norma superior e normalização das gamaglobulinas [8]. No entanto, estudos recentes indicam que a normalização completa das enzimas hepáticas, histologia e gamaglobulinas deve ser o objectivo da terapia, uma vez que esta tem melhorado o prognóstico dos doentes tratados [10, 11].
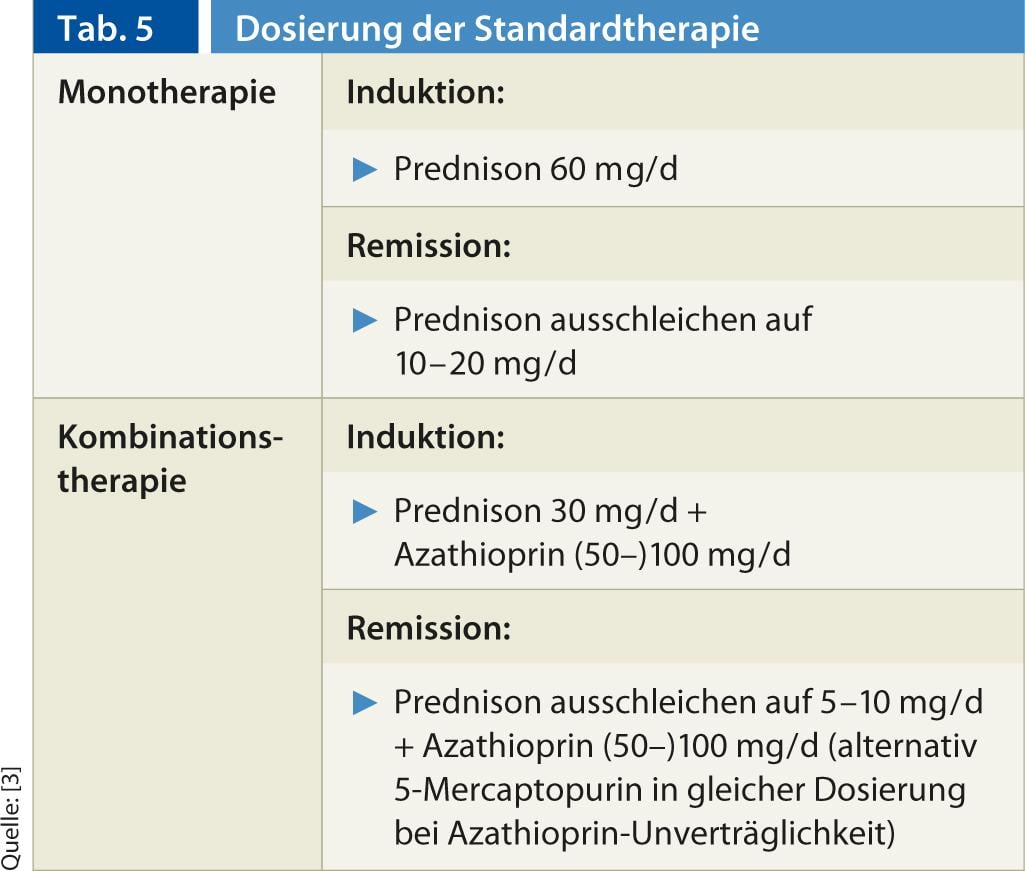
Terapia padrão: Na terapia imunossupressora da AIH, a cooperação do prestador de cuidados primários com um especialista tem provado ser bem sucedida. Actualmente, a terapia padrão consiste em monoterapia ou terapia combinada com prednis(ol)-uma com/sem azatioprina, que se baseia em estudos prospectivos e não aleatórios desde os anos 60 até ao início dos anos 80. Os regimes de dosagem são mencionados no quadro 5. É importante que a dose de esteróides não seja reduzida demasiado depressa. A normalização da transaminase deve ser aguardada. No caso da terapia combinada esteróide/tiopurina, a azatioprina é rapidamente adicionada ao esteroide. Em caso de intolerância à azatioprina, é possível mudar para 5-mercaptopurina (Puri-Nethol) na mesma dosagem. A eficácia da combinação e da monoterapia é comparável, mas a terapia de combinação é a utilização de esteróides, associada a efeitos secundários menos graves (de 66% a <20% durante 18 meses de terapia) [12].
Terapia alternativa com budesonida: No maior estudo de terapia controlada até à data sobre a AIH, o valor da budesonida (3 × 3 mg) foi comparado com a prednis(ol)-uma em cada caso em combinação com azatioprina [13]. Ambos os braços do estudo mostraram uma taxa relativamente baixa de remissões completas, mas significativamente menos efeitos secundários dos esteróides no grupo dos budesonídeos (28% vs. 53%). A budesonida (2 × 3 mg) também parece ser bem adequada para a manutenção da remissão.
No entanto, a hipertensão portal ou cirrose é uma contra-indicação relativa à budesonida devido à redução do metabolismo [14].
Remissão e recaída significante de duração terapêutica suficiente: A remissão é alcançada em 65-75% dos casos após 24 meses de terapia. Cerca de 20% não atingem a remissão completa. A recorrência (aumento das transaminases e/ou sintomas durante a terapia ou durante o afilamento ou após a interrupção da terapia) ocorre em cerca de 50% dos casos no prazo de seis meses e em 80% após três anos após a interrupção da terapia, associada à progressão para cirrose hepática em quase 40% e ao desenvolvimento de insuficiência hepática em 14%.
A duração da terapia antes da cessação da imunossupressão parece ser o principal factor para distinguir os doentes em remissão sustentada dos doentes com recaída. Em comparação com a remissão sustentada de 67% com terapia >4 anos, esta taxa é de apenas 10% com terapia de 1-2 anos [15]. Por conseguinte, recomenda-se a interrupção da terapia apenas com base numa remissão clínica, histológica e bioquímica completa após dois, de preferência quatro anos no mínimo. A remissão histológica pode ser atrasada até oito meses em comparação com a remissão clínica e bioquímica.
Opções terapêuticas mais recentes (“segunda linha”): As opções de tratamento alternativo devem ser discutidas nos seguintes casos:
- Falha no tratamento sob terapia padrão
- Intolerância da terapia padrão
- Evitar os efeitos secundários dos esteróides em doentes de alto risco
- Estudos experimentais
Entre outros, micofenolato, mofetil (MMF), tacrolimus ou ciclosporina A são aqui utilizados.
Transplante hepático: Se a terapia falhar, o transplante hepático deve ser considerado numa fase inicial. Após transplante bem sucedido, a sobrevivência é boa (sobrevivência em 5 anos 91% [16]; sobrevivência em 10 anos 75% [cirrose: 62%]). O risco de recidiva no transplante é de 20-36% [16] e é ainda mais frequente nas crianças.
Monitorização e acompanhamento terapêutico: As transaminases e o nível de IgG indicam o sucesso da terapia e devem ser determinados regularmente (três a seis meses). A determinação de auto-anticorpos clássicos durante o curso não é útil, uma vez que o nível de título não está correlacionado com o grau de actividade. Recomenda-se uma biopsia hepática antes de interromper a terapia, uma vez que uma reacção inflamatória residual no fígado pode já indicar um risco aumentado de recorrência.
Na presença de cirrose, recomenda-se o rastreio do carcinoma hepatocelular. Além disso, os doentes com AIH devem receber a vacinação contra a hepatite A/B, bem como as habituais vacinas padrão [17].
Situações terapêuticas especiais
Pacientes em risco: Os riscos para os efeitos secundários da terapia são: metabolismo diabético, osteoporose, hipertensão arterial mal controlada, capacidade emocional ou história positiva de psicose. Estas situações de risco não são uma contra-indicação absoluta, mas os pacientes devem ser vigiados de perto e as substâncias que contêm esteróides, tais como azatioprina, devem ser utilizadas mais cedo.
Hepatite fulminante: Enquanto um pequeno estudo não demonstrou um benefício relevante dos esteróides na HI fulminante, a resposta dos esteróides em apresentação severa foi de 36-100% noutros estudos. Em geral, o encaminhamento imediato para um centro de transplante é recomendado para apresentação fulminante [18, 19].
Cirrose: Os pacientes com e sem cirrose apresentam um resultado comparativamente bom (morte e transplante de fígado como pontos finais) após dez anos (sobrevivência de 10 anos >90%), após o que a curva de sobrevivência dos pacientes com cirrose (sobrevivência de 20 anos <40%) cai significativamente em comparação com aqueles sem cirrose (sobrevivência de 20 anos <80%).
Cirrose descompensada: Embora o benefício do tratamento não seja claro na cirrose histologicamente inactiva, a resposta do tratamento na cirrose descompensada com hepatite activa pode ser muito boa. Em alguns casos, a regressão da fibrose também é possível [20].
Previsão
Os doentes com AIH sob tratamento adequado têm uma esperança de vida global normal com uma qualidade de vida bem mantida. No entanto, cerca de ¹⁄3 de doentes com AIH já têm cirrose hepática completa no primeiro diagnóstico, com um prognóstico correspondentemente mais pobre. Com excepção dos cursos fulminantes, o transplante de fígado pode ser evitado em quase todos os pacientes não cirróticos e na maioria dos pacientes com cirrose precoce.
Michael Christian Sulz, MD
PD Dr. med. Tilman J. Gerlach
CONCLUSÃO PARA A PRÁTICA
- (AIH) é responsável por 11-23% de todas as hepatopatias crónicas, embora a incidência possa estar subestimada.
- A patogénese exacta da AIH continua a não ser clara. O quadro clínico reflecte uma interacção complexa entre predisposição genética, factores desencadeantes, autoantigénios e mecanismos imunoregulatórios, resultando em última análise num processo imunológico contra os hepatócitos.
- A AIH tem uma apresentação muito heterogénea e flutuante com diferentes manifestações que vão desde a hepatite assintomática à aguda, passando pela insuficiência hepática aguda e cirrose hepática.
- O diagnóstico baseia-se numa combinação de achados clínicos, histológicos e serológicos, bem como na exclusão de outras hepatopatias crónicas. A partitura simplificada de Hennes et al. é fácil de manusear na prática clínica diária.
- A terapia pode ser dividida em diferentes fases tais como indução, manutenção da remissão, afunilamento ou descontinuação e terapia de uma recaída.
- O objectivo da terapia é a remissão permanente; a terapia padrão é a monoterapia ou terapia combinada com prednis(ol)on com/sem azatioprina.
Literatura:
- Boberg KM, et al: Scand J Gastroenterol 1998; 33: 99-103.
- Milkiewicz P, et al: Transplantation 1999; 68: 253-256.
- Manns MP, et al: Hepatology 2010; 51: 2193-2213.
- Yeoman AD, et al: Hepatology 2009; 50: 538-545.
- Czaja AJ, Freese DK.: Hepatology 2002; 36: 479-497.
- Sleisinger, Fordtran’s.: Gastrointestinal e doença hepática. 8ª ed. Vol.2 Saunders, 2006.
- Krawitt EL.: N Engl J Med 2006; 354: 54-66.
- Hennes EM, et al: International Autoimmune Hepatitis Group. Hepatologia 2008; 48: 169-176.
- Czaja AJ, Wolf AM, Baggenstoss AH: Gastroenterologia 1981; 80: 687-692.
- Miyake Y, et al: J Hepatol 2005; 43: 951-957.
- Montano-Loza AJ, Carpenter HA, Czaja AJ.: Am J Gastroenterol 2007; 102: 1005-1012.
- Summerskill WH, et al: Gut 1975; 16: 876-883.
- Manns MP, et al: Gastroenterologia. 2010; 139: 1198-1206.
- Geier A, et al: World J Gastroenterol 2003; 9: 2681-2685.
- Kanzler S, et al: J Hepatol 2001; 34: 354-355.
- Campsen J, et al: Liver Transpl 2008; 14: 1281-1286.
- Gleeson D, Heneghan MA.: Gut 2011; 60: 1611-1629.
- Kessler WR, et al: Clin Gastroenterol Hepatol 2004; 2: 625-631.
- Czaja AJ: Liver Transpl 2007; 13: 953-955.
- Dufour JF, DeLellis R, Kaplan MM: Ann Intern Med 1997; 127: 981-985.
PRÁTICA DO GP 2013; 5: 10-14