Ao lado da hematatose, a fibrose cística (FC) é a doença genética mais comum e a doença genética mais comum com envolvimento pulmonar. Em pacientes adultos, os sintomas são variados e muitas vezes não podem ser distinguidos à primeira vista de outros quadros clínicos. Um historial médico completo e os testes certos ajudam a localizar a doença.
A FC já não é apenas uma doença infantil – 51,2% dos que constam dos registos estão na idade adulta [1]. Na Suíça, cerca de 1000 pessoas são afectadas por isto, lembrou o Dr. Macé Schuurmans, Clínica de Pneumologia, Hospital Universitário de Zurique [2]. CF é herdada de forma autossómica recessiva, havendo aproximadamente 2000 constelações diferentes de alterações hereditárias. O gene afectado codifica uma proteína multifuncional, a CFTR (Cystic Fibrosis Transmembrance Regulator), que serve um canal de cloreto. Existem mutações graves (classe I-III) que estão associadas à insuficiência pancreática. A mutação mais comum que temos na Suíça é mostrada aqui e diz respeito a uma eliminação no local 508 (p.Phe508del) para a fenilalanina neste gene. A insuficiência pancreática surge porque o paciente tem diabetes por um lado, mas por outro lado também um problema digestivo porque as enzimas digestivas não são funcionais.
As terapias moduladoras CFTR estão disponíveis há cerca de 5 anos (ivacaftor, tezacaftor, lumacaftor, elexacaftor). As substâncias individualmente ou em combinação influenciam a função do produto genético, tanto qualitativa como quantitativamente. Esta inovação levou a um grande avanço na terapia da FC, pelo menos para os pacientes com as mutações muito comuns.

Diagnóstico
Desde 2011, tem havido rastreio de recém-nascidos, que é realizado com um teste imunorreactivo de tripsinogénio (IRT) seguido de rastreio de ADN. Normalmente no 4º dia de vida, algumas gotas de sangue são retiradas do calcanhar da criança e colocadas num cartão de papel de filtro para serem analisadas no laboratório de rastreio. Este exame cobre 98% de todas as doenças CF.
As vantagens de testar o mais cedo possível são óbvias: “Sabemos de vários países que introduziram o rastreio mais cedo que o caminho para o diagnóstico é significativamente encurtado”, disse o Dr. Schuurmans. Desta forma, os pais podem ser aconselhados sobre futuras gravidezes, entre outras coisas. A doença manifesta-se menos severamente se for tratada de forma adequada e precoce, e a função pulmonar não diminui tão rapidamente.
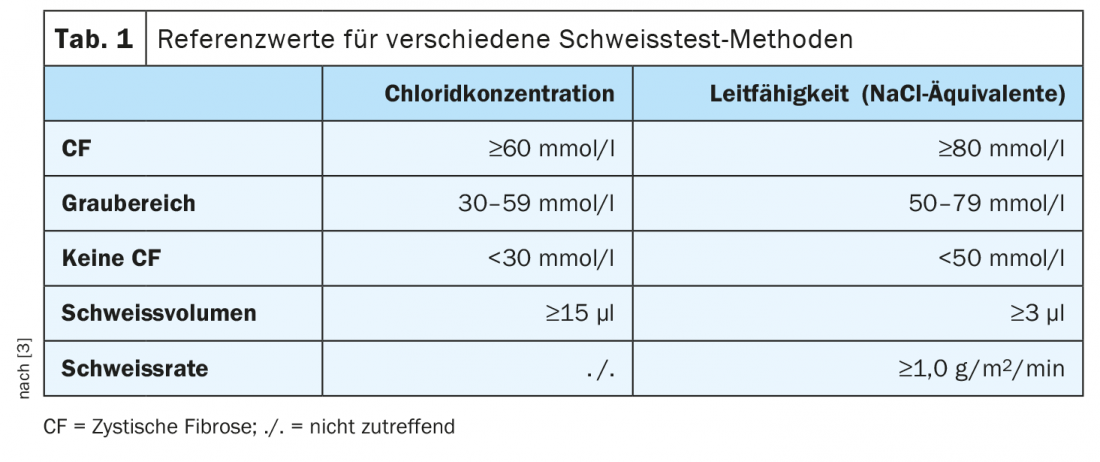
Antes de 2011, este rastreio não estava disponível – todos os indivíduos nascidos antes disso, bem como os adultos que apresentam um diagnóstico tardio, tendem a ter formas mais suaves que não são necessariamente tão nítidas na infância. O primeiro passo mais importante quando se suspeita de FC neste grupo de pacientes é o teste do suor, que é considerado o padrão de ouro para crianças nascidas antes de 2011, bem como para adultos. O teste determina tanto o teor de cloreto como a condutividade. Num caso típico, o teor de cloreto é superior a 60 mmol/l, existe uma gama cinzenta entre 30 e 59 mmol/l. Se não estiver seguro sobre o diagnóstico, pode utilizar a condutividade como ajuda. Se o valor for inferior a 30 mmol/l, por outro lado, é evidente que a FC não está presente (Tab. 1) [3].
Quando pensar em CF
Nos adultos, deve pensar na FC se tiver certos sintomas e também pedir a história da família, aconselha o Dr. Schuurmans. Os sintomas incluem:
- Infecções respiratórias recorrentes, especialmente com certos “germes problemáticos” como o estafilococo. aureus, Pseudomonas aeruginosa, complexo de Burkholderia cepacia ou também micobactérias atípicas (NTM).
- Asma brônquica atípica, isto é, com uma tosse produtiva crónica que não responde à terapia padrão.
- Bronquiectasia manifestando-se antes dos 40 anos de idade. “Isto também pode acontecer mais tarde”, diz o perito. “Mas antes da idade de 40 anos, a exclusão da CF é obrigatória”.
- Polipose nasal, sinusite grave
- Infertilidade nos homens (pode por vezes ser o único sinal nas fases iniciais)
- Perturbação electrolítica (por exemplo, hiponatremia, desidratação)
- Pancreatite aguda
- Doença hepática inexplicada
Se houver suspeita de FC em adultos, recomenda-se primeiro o teste de suor ou o encaminhamento para uma consulta de FC em adultos. O teste do suor é classicamente realizado no hospital infantil. A genética é apenas o segundo passo, e é obrigatório que uma entrevista de aconselhamento seja então realizada por médicos devidamente formados.
Se a fibrose cística não for descoberta até aos 30 anos de idade, então a sobrevivência é correspondentemente mais longa. No entanto, se um paciente for descoberto cedo e a sua função pulmonar for muito boa, então tem significativamente mais anos até que um transplante seja realizado ou o paciente morra, resumiu o Dr. Schuurmans.
Congresso: FomF WebUp Pneumologia
Fontes:
- Zolin A, Orenti A, Naehrlich L, et al: Relatório Anual do ECFSPR 2018.
- FomF WebUp Pneumologia, 7.12.2020.
- Jung A: Schweiz Med Forum 2017; 17(24): 514-522.
HAUSARZT PRAXIS 2021; 16(2): 37 (publicado 19.2.21, antes da impressão).
InFo PNEUMOLOGIA & ALERGOLOGIA 2021; 3(1): 36