
Os critérios de diagnóstico actualmente válidos para a esclerose múltipla (EM) permitem um diagnóstico muito precoce e específico na maioria dos casos. Quando se suspeita de EM, contudo, é sempre importante considerar os diagnósticos diferenciais mais comuns e excluir as “mímicas”. O diagnóstico de síndromes semelhantes à EM tem consequências terapêuticas e, claro, prognósticas. Se surgirem dúvidas sobre o diagnóstico da EM no decurso da doença, não se deve hesitar em renovar a validação do diagnóstico diferencial.
Até à data, não existem marcadores biológicos para o diagnóstico definitivo da esclerose múltipla (EM). Nenhum sintoma neurológico clínico é patognomónico, uma vez que os focos de EM podem ocorrer em qualquer parte do SNC. O tempo médio entre o primeiro sintoma, que muitas vezes não é reconhecido, e o diagnóstico é ainda de cerca de dois anos [1]. Sendo a doença neurológica mais comum da idade adulta jovem, pode levar à incapacidade precoce e à ameaça de reforma antecipada. Tendo em conta os resultados fisiopatológicos e o início precoce da terapia necessária hoje em dia, trata-se de uma perda de tempo que já não pode ser compensada.
A segunda revisão dos critérios McDonald permite que a EM seja diagnosticada no primeiro episódio e com ressonância magnética de sessão única, mas apenas com a exclusão mais cuidadosa possível de outros diagnósticos diferenciais (Tabelas 1 e 2) [2–5].

No sentido de um procedimento de acordo com o princípio de “diagnosticar frequentemente coisas comuns”, os diagnósticos diferenciais (DD) que são postos em causa nas nossas latitudes e no “típico”, ou seja, paciente mais jovem com suspeita de EM, serão tratados com o cursor no seguinte.
Idade e história médica
A distribuição etária da EM mostra um pico de doença por volta dos 30 anos de idade. A esclerose múltipla em crianças e adolescentes e a primeira EM após os 45 anos de idade são menos comuns. Contudo, existem casos individuais bem documentados de EM que se manifestaram pela primeira vez na primeira década de vida, bem como os que se manifestaram na sétima década.
Tomar uma história exacta é essencial e é muitas vezes suficiente para estabelecer um diagnóstico suspeito de EM. A história deve incluir perguntas sobre quaisquer episódios passados de défices neurológicos que possam fornecer pistas sobre recaídas passadas que possam ter sido anteriormente mal interpretadas. É também importante perguntar sobre outras doenças auto-imunes no doente ou em familiares. As perguntas sobre queixas e sintomas na área da bexiga, função rectal e sexual são importantes. Sintomas ocultos como fadiga, problemas de concentração, depressão e dor também devem ser procurados (disfunção autonómica) [3–5]. Os sintomas iniciais comuns da EM são:
- Perturbações de sensibilidade como a formigação, sensação de pele peluda, formigueiro (>30%)
- Distúrbios visuais com visão turva, nebulosa ou nebulosa causados por neurite óptica unilateral (aprox. 16%)
- Perturbações da marcha com fraqueza frequente dependente da carga das pernas e desequilíbrio da marcha (cerca de 5%).
Outros sintomas são: Tonturas (30-50% dos casos), nistagmo (2-4%), tremor intencional, disfunção vesical e intestinal, disfunção sexual, visão dupla, paraestesia facial, neuralgia do trigémeo, ataxia, disartria, fadiga (especialmente em caso de recaída), distúrbios do sono, depressão, euforia, disfunção cognitiva (34-65%), demência (5%), convulsões cerebrais (2-3%) e sinal positivo de Lhermitte.
Diagnóstico diferencial da EM
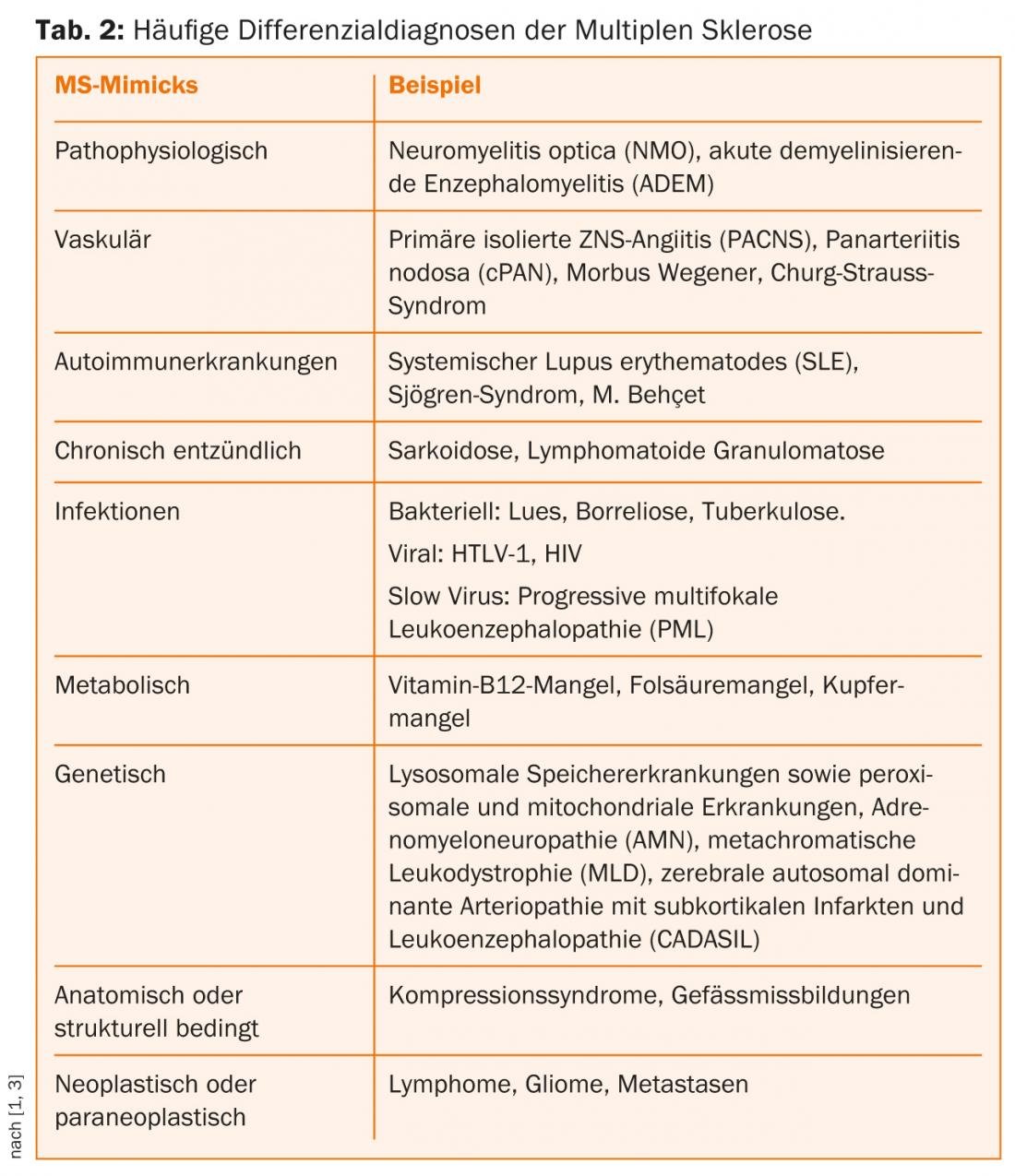
A riqueza das formas dos sintomas da EM torna tão difícil diferenciá-las de toda uma gama de afecções cerebroespinais. Outras doenças crónicas inflamatórias, metabólicas, vasculares, neoplásicas e/ou mediadas por agentes patogénicos podem apresentar-se com sintomas semelhantes (Tab. 2) [3–5].
Fisiopatologia Mimicks
A EM é uma doença inflamatória crónica imunomediada do sistema nervoso central que, histopatologicamente, leva a vários graus de dano axonal e desmielinização. Há várias outras doenças auto-imunes que envolvem o sistema nervoso central e que são difíceis de distinguir da EM devido à variedade de sintomas.
A neuromielite óptica (NMO) é também uma doença inflamatória crónica, imuno-mediada do SNC. As “doenças do espectro NMO” (NMOSD) incluem mielite transversal extensiva longitudinal isolada (LETM), neurite óptica isolada monofásica ou recorrente e formas distintas de encefalite do tronco encefálico. NMO mostra uma clara preferência pelo sexo feminino (até 9:1 dependendo da coorte) e manifesta-se em média cerca de dez anos mais tarde (4ª década de vida) do que a EM. Devido à detecção de anticorpos aquaporina-4 (AQP4-Ak), que são dirigidos contra os canais de água expressos em astrocitos, a NMO foi classificada como uma doença autoimune mediada por células B. A detecção serológica de AQP4-Ak e a contagem de células predominantemente linfocitárias aumenta de >20 a 50 µl no LCR não são incomuns no NMO no contexto de uma recidiva aguda. Além disso, as bandas oligoclonais positivas só são encontradas em cerca de 15-30% dos doentes com NMO. Em comparação com a EM, as recaídas em NMO são frequentemente mais graves e conduzem mais rapidamente a incapacidades permanentes devido a uma remissão na sua maioria incompleta [5].
A encefalomielite desmielinizante aguda (ADEM) e a sua variante máxima, a leucoencefalite hemorrágica aguda (AHLE), são doenças raras e inflamatórias desmielinizantes do SNC. Ao contrário da EM, a ADEM é mais comum na infância do que nos adultos e ocorre principalmente em estreita relação temporal com infecções ou vacinações. Não existem critérios de diagnóstico claramente definidos para a ADEM. Os raros casos de adultos são um desafio [5].

Angite e colagenoses
A angite primária isolada do SNC (PACNS) é uma doença auto-imune muito rara caracterizada por danos inflamatórios em vasos cerebrais de pequeno e médio calibre. Pode ser considerado como um diagnóstico diferencial na presença de sintomas multifocais ou difusos do SNC com um curso progressivo ou recorrente. A ressonância magnética e a angiografia de subtracção digital fornecem pistas. Se alterações vasculíticas estiverem presentes fora do CNS, o PACNS é excluído [5].
Os vasculídeos (primários) sistémicos são classificados de acordo com o tamanho dos vasos afectados e a sua constelação histológica de descobertas. Panarterite nodosa (cPAN), doença de Wegener e síndrome de Churg-Strauss são diagnósticos diferenciais possíveis da EM. Caracterizam-se por inflamação granulomatosa e necrosante dos pequenos vasos e pela presença de anticorpos antinucleares (ANCAs).
Doenças auto-imunes como o lúpus eritematoso sistémico (LES), a síndrome de Sjögren ou a doença de Behçet raramente podem causar sintomas neurológicos devido à vasculite. Em princípio, em todas as doenças inflamatórias crónicas auto-imunes, hemorragias e enfartes isquémicos na área de abastecimento das áreas vasculares afectadas podem complicar o curso e conduzir a sintomas neurológicos graves no SNP e SNC (vasculites secundários).
A doença de Behçet é uma vasculite sistémica e afecta predominantemente os homens da região mediterrânica. O pico da doença ultrapassa a terceira década de vida, e a primeira manifestação raramente ocorre na adolescência. Encontra-se uma associação notoriamente frequente com o antigénio HLA-B51 nas pessoas afectadas. Ulcerações orais e genitais recorrentes e alterações cutâneas sob a forma de eflorescências pustulopapulares estéreis e eritema nodoso são típicas. O envolvimento dos olhos é típico. Raramente existe uma manifestação parenquimatosa do SNC (meningoencefalite), que afecta o tronco encefálico em cerca de 50% dos casos. O aspecto neurológico varia em função da área afectada do cérebro ou das estruturas vasculares afectadas e pode ser tanto em recidiva como em progressão crónica. O Neuro-Behçet é detectado com ressonância magnética e diagnóstico do QCA [5].
Em doenças granulomatosas como a neurosarcoidose, o envolvimento do SNC por si só é extremamente raro; o co-envolvimento é encontrado em cerca de 5% dos doentes com sarcoidose. Nas manifestações do SNC, o envolvimento do nervo craniano na sua maioria unilateral (nervo facial, nervo óptico, nervo acústico) é o acontecimento inicial mais comum. A aparência clínica adicional depende essencialmente da localização e extensão dos granulomas no SNC.
Causas infecciosas
Embora as neuroligas fossem as DD mais comuns da EM em 1925, são agora raras e as novas causas infecciosas são mais importantes. Os sintomas neurológicos de lues causados por Treponema pallidum podem ser descritos como meningite precoce (fase 2), lues cerebrospinalis com arterite cerebral concomitante (fase 3) e paralisia progressiva/tabes dorsalis (fase 4) fazer uma aparição. Especialmente na fase 3, os sintomas variam muito. Focos desmielinizantes no cérebro e medula espinal caracterizam a fase de tabes dorsalis [5].
Em termos de diagnóstico diferencial, a neuroborreliose deve ser considerada, especialmente em regiões com uma elevada taxa de infestação de carraças com Borrelia burgdorferi, mesmo que a sua frequência em pessoas infectadas com Borrelia seja baixa a 0,3-1,4%. Distinguem-se três fases, em que a fase 1, o eritema migrante local, ainda não apresenta quaisquer sintomas neurológicos. Se não for tratada, uma infecção disseminada (fase 2) pode desenvolver-se após semanas a meses, envolvendo vários sistemas de órgãos e, portanto, também o CNS e o PNS. A neuroborreliose aguda caracteriza-se por dores radiculares segmentares, muitas vezes analgésico-resistentes, sem sinais meníngeos de irritação. No decurso da doença, mais de 50% dos doentes desenvolvem irritações sensoriais, paresia das extremidades e défices do nervo craniano (nervo facial, nervo raptado). Após meses a anos, podem desenvolver-se sintomas no sentido de neuroborreliose crónica progressiva (fase 3: mielite, miosite, vasculite e polineuropatia). A RM desempenha um papel subordinado na neuroborreliose; as provas de alterações inflamatórias do LCR e a produção de anticorpos específicos de Borrelia intratecal são aqui conclusivas.
Entre os vírus, deve ser mencionado o vírus da leucemia de células T humanas 1 (HTLV-1), um retrovírus patogénico humano que infecta predominantemente células T CD4+ e, em casos muito raros, causa leucemia de células T e/ou conduz a sintomas neurológicos. A paraparesia espástica tropical é uma doença muito rara nas nossas latitudes, que tem principalmente um curso espinal. A infecção pelo VIH também pode causar encefalomielite nas suas fases avançadas.
Causas metabólicas
A mielose funicular é uma doença com deficiência de vitamina B12 que leva à desmielinização do cérebro traseiro, cerebelar sidebrain e vias piramidais, especialmente na medula cervical e torácica. Semelhante à deficiência de vitamina B12, as perturbações do metabolismo do ácido fólico podem levar a uma variedade de sintomas em diferentes graus de gravidade. As perturbações neurológicas incluem convulsões, atrasos de desenvolvimento e sintomas mielopáticos semelhantes aos da mielose funicular. As doenças por deficiência vitamínica são raras nas nossas latitudes (álcool, desnutrição). Ainda mais raros são os distúrbios de utilização de vitaminas devido a doenças genéticas.
Causas genéticas
Em casos raros, as doenças monogénicas podem também ter semelhanças fenotípicas com a manifestação clínica da EM. Estas doenças hereditárias incluem doenças de armazenamento lisossomal, bem como doenças peroxisomais e mitocondriais, tais como a atrofia óptica de Leber. Uma vez que na maioria dos casos as manifestações clínico-neurológicas já ocorrem na infância e infância, os neuro-pediatras são chamados a intervir.
A adrenoleucodistrofia ligada ao X (ALD) é uma doença de armazenamento peroxisomal caracterizada por degradação deficiente dos ácidos gordos de cadeia longa. Também começa normalmente na infância, mas também há uma forma adulta muito rara, a adrenomieloneuropatia (AMN: paraparesia espástica, insuficiência adrenal).
Na forma adulta de leucodistrofia metacromática (MLD), uma das 30 diferentes doenças de depósito lisossómico, distúrbios da marcha espástica, incontinência e, mais raramente, atrofia óptica e distúrbios do movimento ocular podem ocorrer, para além dos sintomas neuropsicológicos frequentes. O factor decisivo na diferenciação clínica da EM no MLD é o envolvimento do sistema nervoso periférico sob a forma de uma polineuropatia sensorimotora acentuada distalmente, na sua maioria simétrica.
Paraneoplasias e neoplasias
Os pacientes com sintomas neurológicos têm frequentemente muito medo de sofrer de um tumor cerebral ou da medula espinal ou de metástases. Um curso de recaída com tendências de regressão fala contra ele. Dor de cabeça, enjoos matinais, vómitos, alterações de personalidade, abrandamento e ataques epilépticos podem ocorrer tanto em lesões malignas como benignas do espaço intracraniano, mas são raras na EM.
Suspeita de um diagnóstico errado?
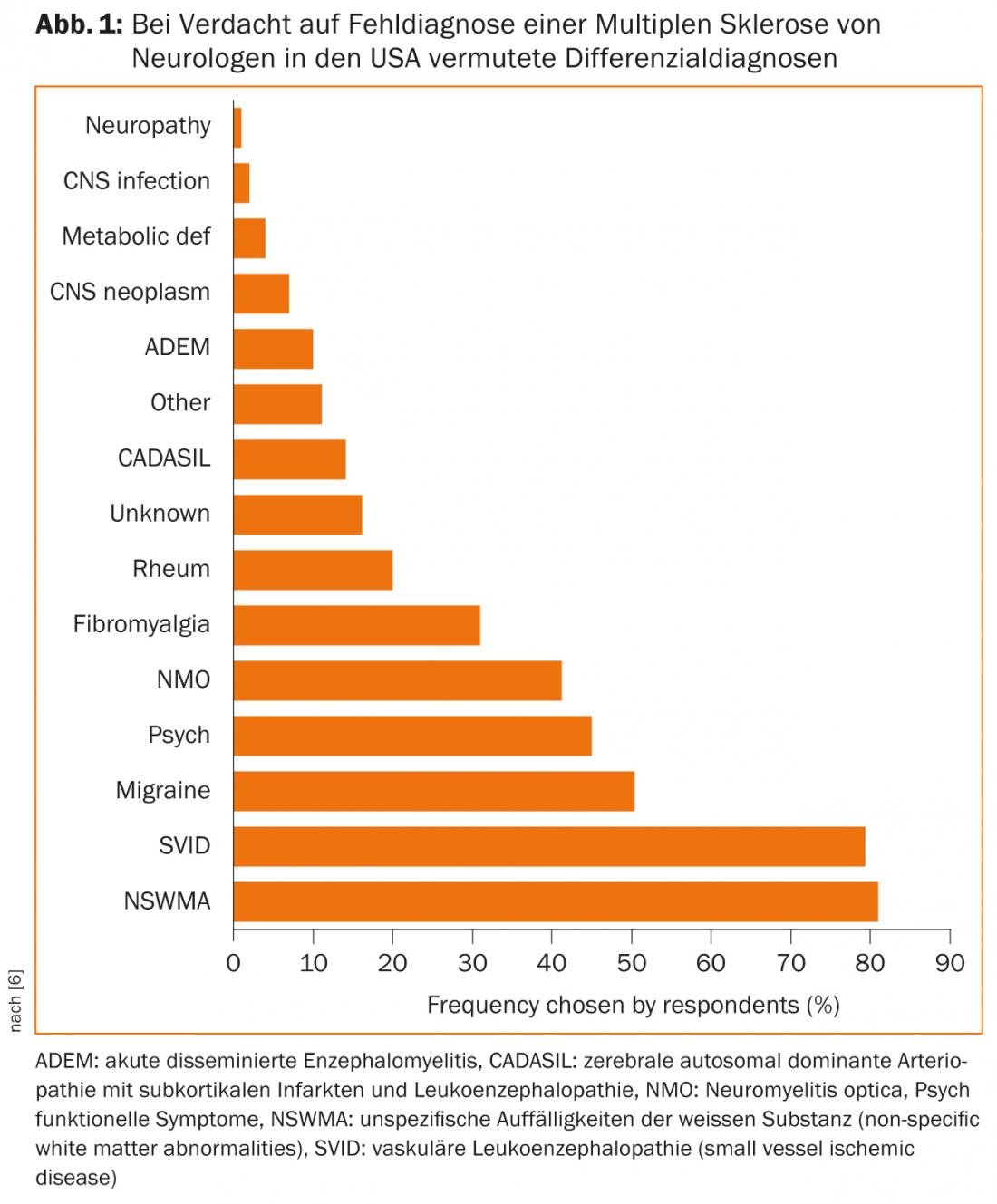
Em casos com um curso atípico, falta de resposta às opções de tratamento actuais ou sintomas fulminantes, é urgente rever o diagnóstico diferencial e, se necessário, consultar peritos de outras disciplinas, tais como reumatologistas e infectologistas. Um inquérito aos neurologistas americanos mostra quais os DD que são frequentemente suspeitos quando se suspeita de um diagnóstico incorrecto (Fig. 1) [6].
Prof. Adam Czaplinski, MD
Literatura:
- Fernández O, et al: Características da esclerose múltipla no início e atraso do diagnóstico e tratamento em Espanha (o Novo Estudo). J Neurol 2010; 257(9): 1500-1507.
- Polman CH, et al: Critérios de diagnóstico para esclerose múltipla: revisões de 2010 dos critérios McDonald. Ann Neurol 2011; 69: 292-302.
- Marcus JF, et al: Updates on Clinically Isolated Syndrome and Diagnostic Criteria for Multiple Sclerosis (Actualizações sobre Síndrome Clinicamente Isolada e Critérios de Diagnóstico para Esclerose Múltipla). Neurohospitalista 2013; 3(2): 65-80.
- www.awmf.de: Esclerose múltipla DGN número de registo AWMF: 030/050.
- Décard BF, et al: Diagnóstico diferencial de esclerose múltipla (EM) – Mímicas de EM. Lei Neurol 2012; 39: 83-99.
- Solomon AJ, et al: Undiagnosing MS: The Challenge of misdiagnosis in MS. Neurologia 2012; 78; 1986-1991.
InFo Neurologia & Psiquiatria 2013; 11(6)









