No passado, um diagnóstico de fibrose cística (FC) era sinónimo de morte precoce, geralmente antes dos 30 anos de idade. A doença hereditária ainda está a encurtar a vida, mas graças aos avanços na investigação e novas terapias, a esperança média de vida na Europa Central já foi aumentada para mais de 50 anos. Para as pessoas afectadas na idade adulta, há agora uma série de opções para gerir a fibrose cística.
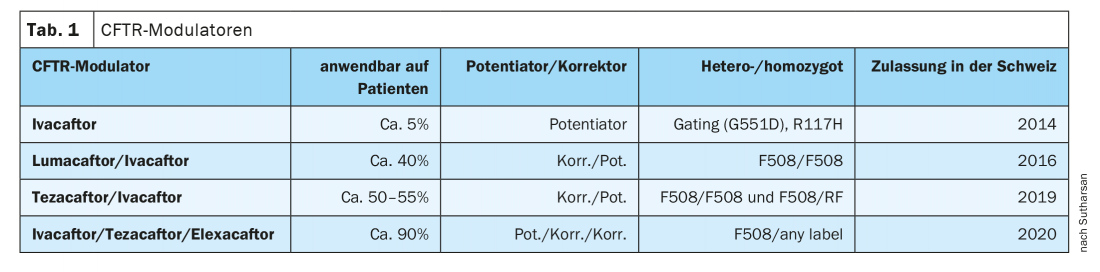
A introdução de enzimas pancreáticas foi um grande avanço terapêutico, mas o estabelecimento de centros onde os doentes com FC recebem tratamento específico também contribuiu para aumentar a probabilidade de sobrevivência, disse o Dr. Sivagurunathan Sutharsan, médico sénior da Clínica de Pneumologia, Clínica Ruhrland, Clínica de Medicina Universitária Essen (D), a título de introdução. Em 1989, foi descoberto o gene CFTR. Embora ainda não exista terapia genética, a medicina personalizada também chegou em fibrose cística, diz o especialista: em 2014, o ivacaftor (IVA) foi aprovado na Suíça como a primeira substância activa para um pequeno grupo de doentes (5-6%); entretanto, existem mais três moduladores que estão disponíveis como combinações e, desde Agosto de 2020, também como um triplo (Tab.1).
Complicações crescentes com a idade
As primeiras complicações já são visíveis na infância: as crianças apresentam frequentemente (até 25%) o íleo de mecónio como o indicador mais antigo, geralmente (>85%), a insuficiência pancreática já está presente e as infecções pulmonares ocorrem frequentemente (no entanto, apenas <25% Pseudomonas aeruginosa). Durante a infância e a adolescência, a proporção de P. aeruginosa aumenta para aproximadamente 45%, mas outros órgãos também entram em jogo, incluindo doenças hepáticas e pólipos nasais (25%). Na idade adulta, a doença completa aparece finalmente, incluindo doenças pulmonares e ósseas graves (visão geral 1).
Durante a primeira década de vida, Staphylococcus aureus e Haemophilus influenzae são as bactérias mais comuns isoladas da expectoração na FC; na segunda e terceira décadas de vida, Pseudomonas aerugiosa é muito mais comum.

As exacerbations aumentam o risco
A gestão da fibrose cística é fundamental para evitar exacerbações. “Os pacientes que têm mais de 2 exacerbações pulmonares por ano estão em risco acrescido de morte ou transplante pulmonar”, explicou o Dr. Sutharsan. A redução do número de exacerbações e subsequente perda da função pulmonar deve, portanto, ser um objectivo importante da gestão da FC.
Na gestão, a fisioterapia é geralmente prescrita para retirar as secreções dos pulmões, a antibioticoterapia é indicada para infecções crónicas com Pseudomonas ou Staphylocococci, e macrólidos como a azitromicina são frequentemente utilizados como terapia a longo prazo para a colonização crónica de Pseudomonas. Se os doentes estiverem a sangrar, existe a opção de realizar a oclusão da artéria brônquica. A terapia com moduladores CFTR é nova (visão geral 2).

Moduladores CFTR
Na mutação mais comum, F508del, a proteína é produzida, mas é normalmente degradada na célula. Mas com os novos moduladores, os correctores CFTR elexacaftor (ELX) e tezacaftor (TEZ), o canal de cloro é estabilizado em diferentes locais de ligação, evitando assim a degradação. Segue-se um transporte directo para a superfície da célula, onde o potenciador CFTR ivacaftor pode aumentar a probabilidade de abertura do canal de cloreto. Existem agora 4 moduladores CFTR aprovados. Enquanto o ivacaftor pode ser utilizado em cerca de 5% da população de FC, a nova combinação tripla de ivacaftor/tezacaftor/elexacaftor pode tratar cerca de 90% dos pacientes em todo o mundo. Como pré-requisito para o tratamento, deve-se ter pelo menos uma mutação 508 num alelo. “A segunda mutação normalmente não importa”, diz o Dr. Sutharsan. Os principais efeitos secundários descritos são erupções cutâneas e prurido, que são geralmente bem controlados com anti-histamínicos orais ou esteróides. Contudo, um bom efeito é descrito não só nos pulmões, mas também nos seios nasais.
Na sua clínica em Essen, o Dr. Sutharsan e os seus colegas ajustaram 133 pacientes CF ao novo triplo ELX/TEZ/IVA (80 homozigotos, 32 + 21 (caso de dificuldade) heterozigotos). No grupo homozigotos, houve 96 hospitalizações com terapia i.v. devido a exacerbações nos 12 meses anteriores. Em 6 meses sob o triplo, este número foi reduzido para apenas 5 terapias i.v. A situação era semelhante nos heterozigotos: 29 + 74 (dificuldade) tratamentos i.v. antes do início durante um período de 12 meses tornaram-se apenas 2 + 7 (dificuldade) tratamentos i.v. nos primeiros 6 meses sob ELX/TEZ/IVA. O rumo seguinte ainda tem de ser visto, mas os números “já mostram o enorme progresso na terapia que os novos moduladores trazem consigo”, concluiu o Dr. Sutharsan.
Fonte: 128. Congresso da Sociedade Alemã de Medicina Interna (DGIM)
InFo PNEUMOLOGIA & ALERGOLOGIA 2022; 4(2): 30-31