Os anti-histamínicos H1 estão divididos numa primeira geração, mais antiga, com efeitos secundários sedantes e numa segunda geração sem estes efeitos. O nosso autor lança luz sobre a farmacologia deste grupo de substâncias activas.
Até agora, são conhecidos quatro receptores histamínicos diferentes. Estas moléculas acopladas à proteína G estão localizadas na superfície celular e exercem efeitos diferentes, dependendo do local de expressão. Enquanto a activação do receptor H1 leva em particular ao prurido, vasodilatação, contracção da musculatura lisa com broncoespasmo ou cólicas abdominais, secreção de muco com rinorreia e aumento da secreção brônquica bem como um aumento da permeabilidade vascular, os receptores H2 estão envolvidos em particular no aumento do suco gástrico e da secreção ácida. Além disso, existem receptores H3, que desempenham um papel no CNS como autorreceptores pré-sinápticos, e receptores H4, que desempenham um papel na diferenciação e modulação das células imunitárias, entre outras coisas. Neste artigo, as substâncias dirigidas contra o receptor H1 são discutidas, enquanto que os antagonistas dos receptores H2, dos quais apenas a ranitidina ainda está no mercado na Suíça, não são tratados. Agonistas e antagonistas nos receptores H3 e H4 estão em desenvolvimento clínico.
Farmacodinâmica dos anti-histamínicos H1
Na Suíça, estão disponíveis 22 substâncias activas da classe anti-histamínica H1. Actualmente, assume-se que os anti-histamínicos estabilizam o receptor H1 na sua conformação inactiva e assim asseguram que menos receptores podem ser activados pela histamina. Enquanto os anti-histamínicos H1 da primeira geração mais velha penetram bem no sistema nervoso central e aí exercem um efeito sedativo nos receptores H1 pós-sinápticos, o mesmo não acontece com os representantes da segunda geração ou não em concentrações terapêuticas (Tab. 1) . Devido à sua boa eficácia nos receptores centrais H1, alguns representantes da Sedativos/hipnóticos de primeira geração (ex. doxilamina, difenidramina), antieméticos (ex. meclozina) ou contra o enjoo do movimento (ex. dimenidrinato). A fraca penetração do SNC pelos representantes do A segunda geração deve-se ao facto de estas substâncias serem hidrofílicas e substratos da glicoproteína P do transportador dirigido para o exterior, presentes na barreira hemato-encefálica (entre outras barreiras de membrana do corpo). Isto evita a sedação, que é utilizada em indicações anti-alérgicas com substâncias do A 1ª geração era muitas vezes limitadora da terapia. Alguns anti-histamínicos H1 do A 1ª geração tem efeitos adicionais nos receptores de acetilcolina, noradrenalina e serotonina, enquanto os representantes da A 2ª geração inactiva especificamente o receptor H1.
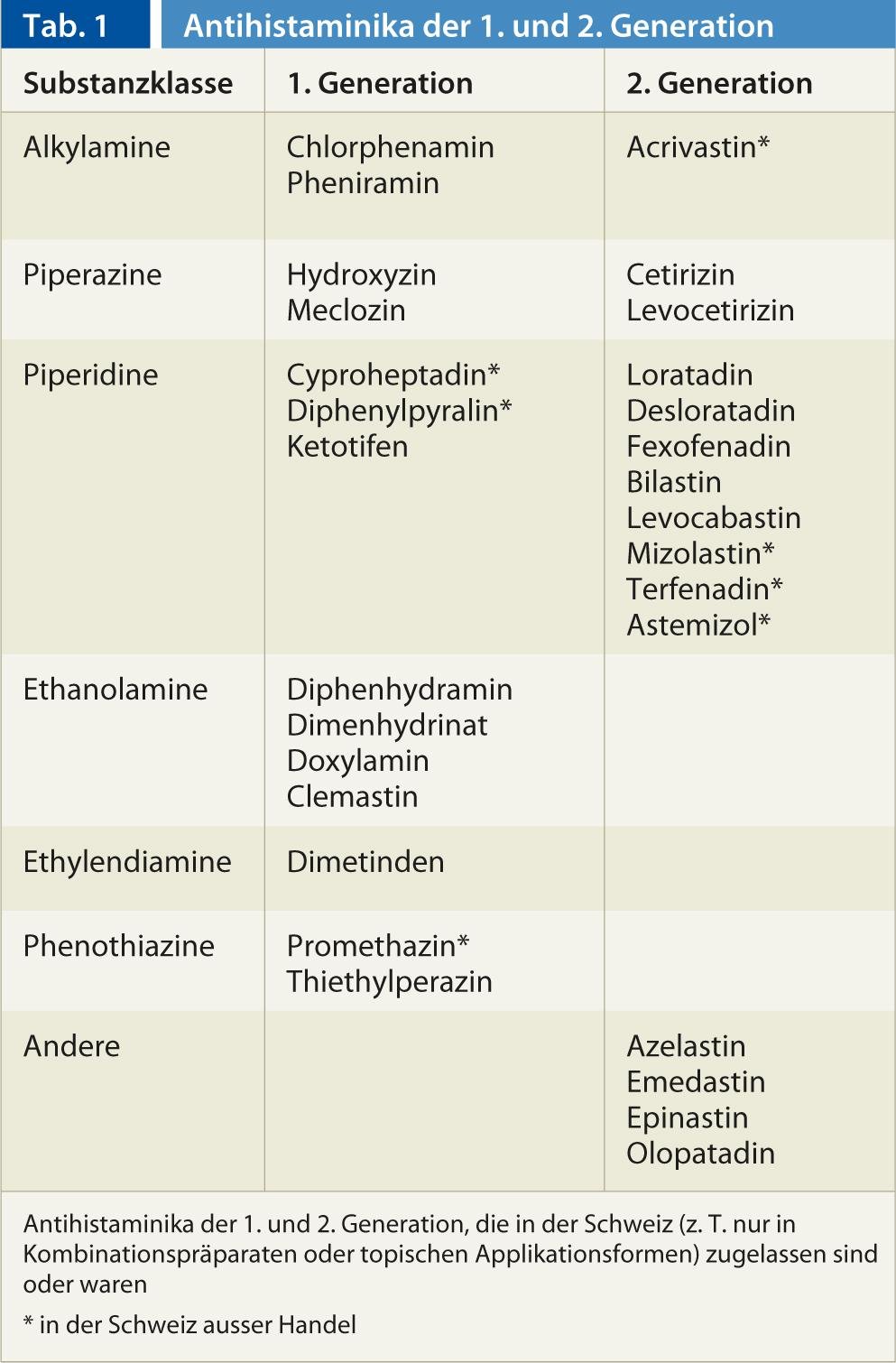
Globalmente, a eficácia clínica dos anti-histamínicos H1 do 1. geração têm sido mal estudadas em ensaios clínicos, enquanto as provas para o uso de anti-histamínicos H1 da A segunda geração é boa para a rinite alérgica, conjuntivite alérgica e urticária. A utilização de produtos da A segunda geração em dermatite atópica, asma, anafilaxia, angioedema não alérgico, constipações, prurido de origem não alérgica, etc., foi mal investigada em estudos ou os estudos não mostraram efeitos convincentes e também não há aprovação para tais indicações. Nas crianças, as substâncias do A 1ª geração pode levar a efeitos secundários por vezes ameaçadores, pelo que a indicação deve ser feita com especial cuidado. Numa chamada 3ª geração, os enantiómeros ou metabolitos das moléculas do 2ª geração, sem que haja grandes diferenças farmacodinâmicas, de modo a que as substâncias sejam farmacologicamente semelhantes ao 2ª geração.
Farmacocinética de anti-histamínicos H1
O primeiro obstáculo que um medicamento tem de ultrapassar para ser eficaz é a sua ingestão. A garantia do cumprimento ou da adesão é, portanto, de particular importância na prática médica diária. A maioria dos anti-histamínicos H1 para uso sistémico estão disponíveis como formas de dosagem sólida (comprimidos, drageias, comprimidos revestidos por película, supositórios) ou gotas para administração oral. Algumas substâncias são aplicadas localmente (por exemplo, gotas para os olhos). Apenas alguns podem também ser administrados por via intravenosa (dimetinden, clemastine, thiethylperazine). A reabsorção, que é rápida para a maioria dos anti-histamínicos de segunda geração H1 e leva a níveis máximos após uma a três horas, é seguida de distribuição no sangue e tecidos, metabolismo se necessário, e excreção.
Existem grandes diferenças entre os anti-histamínicos, especialmente na metabolização e eliminação (Tab. 2). Além disso, existem diferenças no metabolismo entre pessoas causadas por influências genéticas e ambientais. Em particular, a enzima citocromo P450 CYP2D6, que está envolvida na degradação de alguns anti-histamínicos H1 (Tab. 2) , mostra uma forte variabilidade genética, que pode variar desde a ausência até à multiplicação da actividade enzimática normal. Além disso, o CYP2D6 pode ser inibido por algumas substâncias: bupropion, cinacalcet, os inibidores selectivos de recaptação de serotonina paroxetina, fluoxetina e duloxetina, bem como o medicamento antifúngico terbinafina estão entre os inibidores mais fortes do CYP2D6. Contudo, uma vez que todos os anti-histamínicos H1 são degradados por mais de uma enzima e são frequentemente eliminados renalmente inalterados, a variabilidade genética e a inibição do CYP2D6 muito raramente desempenham um papel na utilização de anti-histamínicos H1.
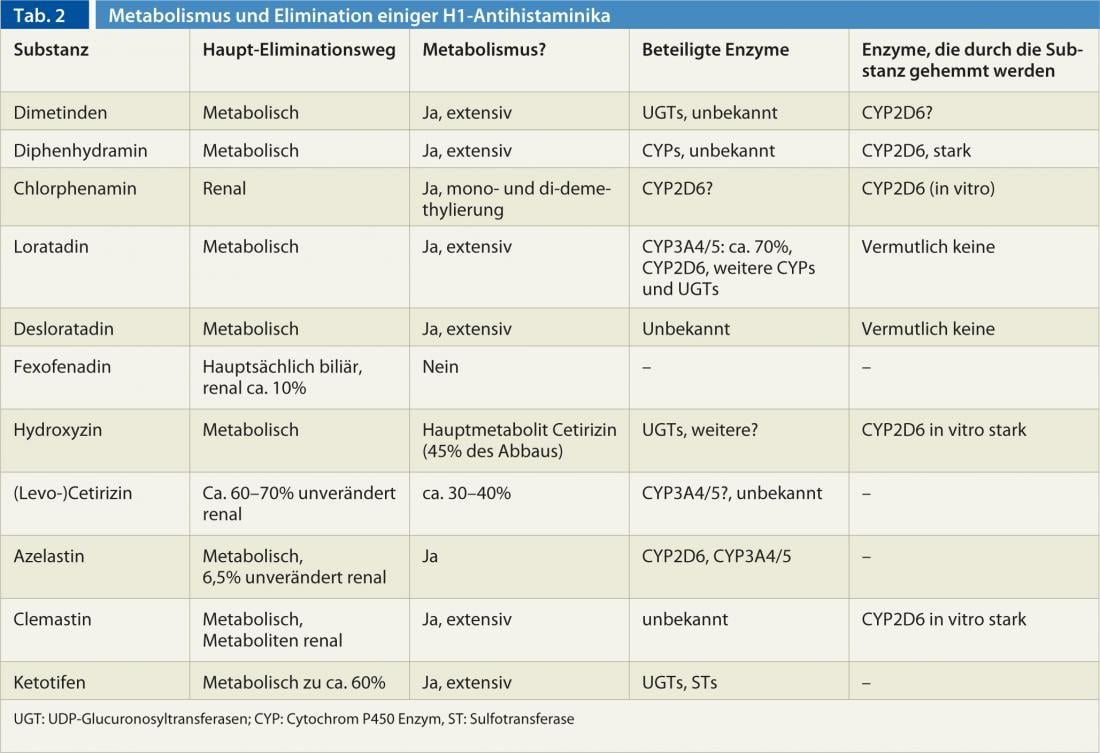
Alguns anti-histamínicos de 1ª geração H1 são também inibidores do CYP2D6. (Tab. 2), para que quando os substratos de CYP2D6 (por exemplo codeína, dextrometorfano, muitos antipsicóticos [haloperidol, risperidona, aripiprazole], atomoxetina, muitos antidepressivos [a maioria dos tricíclicos, venlafaxina, etc.], bem como os beta-bloqueadores metoprolol, carvedilol e timololol) sejam utilizados concomitantemente, uma dose mais baixa do substrato deve ser escolhida para evitar efeitos adversos.
Globalmente, a farmacocinética das substâncias da 1ª geração só é conhecida de forma incompleta. é apenas incompletamente conhecida, uma vez que as substâncias foram aprovadas há décadas com muito menos dados do que os que são necessários hoje em dia. Mas também há lacunas no conhecimento sobre os preparativos da segunda geração: A desloratadina é metabolizada ao metabolito igualmente activo 3-hydroxy-desloratadine, mas a enzima ou enzimas envolvidas são desconhecidas, embora se tenha verificado que 2% dos europeus e até 20% dos africanos não podem formar 3-hydroxy-desloratadine. As enzimas CYP2D6 e CYP3A4/5 envolvidas no metabolismo da loratadina não parecem ser responsáveis por isto.
O CYP3A4 é a mais importante enzima citocromo P450, tanto em termos de quantidade como de número de substratos. Embora não sejam conhecidas variantes genéticas que alterem a função do CYP3A4 (apesar da investigação intensiva), existem medicamentos que aumentam a actividade enzimática (rifampicina, efavirenz, fenitoína, carbamazepina, ingredientes da erva de São João, etc.) e a inibem (antifúngicos azole, eritromicina, claritromicina, ritonavir, verapamil, diltiazem, amiodarona, etc.).(antifúngicos azole, eritromicina, claritromicina, ritonavir, verapamil, diltiazem, amiodarona, ingredientes de toranja, especialmente em sumo de toranja, etc.). O CYP3A5, por outro lado, que decompõe essencialmente as mesmas drogas que o CYP3A4, quase nunca está presente nos europeus devido a uma variante genética, enquanto que é normalmente funcional nos africanos. Com excepção da loratadina, azelastina e provavelmente também cetirizina, o CYP3A4/5 não desempenha um papel nos anti-histamínicos.
Foi ainda mais surpreendente quando um efeito do sumo de toranja foi demonstrado num estudo com fexofenadina: Quando a fexofenadina foi tomada juntamente com sumo de toranja, os níveis de fexofenadina diminuíram, especialmente pouco tempo depois de a tomar, em comparação com a tomada com água (um aumento teria sido esperado se o CYP3A4 fosse inibido pelas furanocumarinas do sumo de toranja). Este efeito pode ser explicado pelo facto de outras substâncias no sumo de toranja (o flavonóide naringin) inibirem o intestino, transportador interno OATP1A2, que é necessário para a absorção da fexofenadina do lúmen intestinal.
Outra interacção que ainda não foi totalmente esclarecida diz respeito à fexofenadina: a administração simultânea de itraconazol levou a níveis muitas vezes superiores de fexofenadina. Uma vez que a fexofenadina não é metabolizada pelo CYP3A, que é potentemente inibido pelo itraconazol, a interacção foi explicada pela inibição da P-glycoprotein, o transportador que se pensa desempenhar um papel importante na eliminação da fexofenadina. Se esta hipótese é correcta e outros antifúngicos azóicos também levam a aumentos de nível ainda precisam de ser investigados.
Por outro lado, é de notar que substâncias como a (levo-)cetirizina são principalmente eliminadas renalmente, pelo que as restrições na função renal devem também levar a reduções de dose. Para a levocetirizina, por exemplo, é necessário reduzir a dose a partir de um taxa de filtração glomerular inferior a 50 mL/min, é prescrita uma administração de 5 mg a cada 2º dia; em caso de comprometimento mais grave da função renal, o intervalo de dosagem deve ser ainda mais alargado.
Na Suíça, é possível obter informações relacionadas com os efeitos secundários e interacções, ajustamentos de dose e outros problemas relacionados com drogas nas instalações de farmacologia clínica dos hospitais universitários.
Efeitos adversos dos anti-histamínicos: Foco QTc tempo
Embora a maioria dos efeitos adversos dos anti-histamínicos H1 se devam à sua acção no recetor H1 (fadiga, redução do desempenho cognitivo e psicomotor, aumento do apetite) ou (no caso das substâncias mais antigas) por efeitos no receptor de m-acetilcolina (boca seca, retenção urinária, taquicardia), no receptor de alfa-adrenocepção (hipotensão, tonturas, taquicardia reflexa) ou no receptor de serotonina (por exemplo, aumento do apetite). Embora os efeitos dos anti-histamínicos H1 possam ser explicados pelos seus efeitos no receptor alfa-adrenérgico (hipotensão, tonturas, batimentos cardíacos reflexos) ou no receptor de serotonina (por exemplo, aumento do apetite), é menos conhecido que alguns anti-histamínicos H1 também inibem os canais de iões cardíacos, especialmente o canal IKr, que modula a rápida saída de potássio durante a repolarização e pode assim levar a um prolongamento da repolarização (do intervalo QT no ECG) e mesmo a torsades de pointes de taquicardia ventricular.
Globalmente, o potencial dos anti-histamínicos H1 para prolongar o tempo de QTc no ECG é mal estudado. O interesse por este efeito secundário potencialmente fatal foi despertado quando houve vários relatórios de torsades de pointes episódios para a primeira segunda geração de anti-histamínicos H1, terfenadina e astemizol. Na maioria dos casos, tinham sido tomadas overdoses ou não tinham sido observadas interacções que aumentassem a concentração. As duas substâncias foram retiradas do mercado em 1990 devido a arritmias ventriculares. Para a loratadina, fexofenadina e cetirizina, existem relatórios de casos individuais de prolongamento do tempo QTc e, em alguns casos, torsades de pointes tachycardia.
Os factores de risco gerais para o prolongamento do tempo QTc são apresentados no Quadro 3 . Por conseguinte, parece aconselhável, especialmente em doentes de risco a quem são administrados anti-histamínicos regularmente e em doses elevadas, tomar um ECG e verificar o soro de potássio e magnésio.

CONCLUSÃO PARA A PRÁTICA
- Os anti-histamínicos H1 da 2ª geração são geralmente bem tolerados.
- O prolongamento do tempo QTc é possível, especialmente com overdoses e factores de risco.
- O sumo de toranja não deve ser bebido com fexofenadina, pois reduz as concentrações de fexofenadina. O itraconazol aumenta as concentrações de fexofenadina, o que pode levar a sintomas de overdose.
- No caso de anti-histamínicos principalmente renalmente excretados, como a (levo-)cetirizina, a dose deve ser ajustada em caso de insuficiência renal.
PD Alexander Jetter, MD
Literatura:
- Simons FE, Simons KJ: Histamina e H1-anti-histamínicos: celebrando um século de progresso. J Allergy Clin Immunol 2011; 128: 1139-1150.
- Shon JH, Yoon YR, Hong WS, Nguyen PM, Lee SS, Choi YG, Cha IJ, Shin JG: Efeito do itraconazol sobre a farmacocinética e farmacodinâmica da fexofenadina em relação ao polimorfismo genético MDR1. Clin Pharmacol Ther 2005; 78: 191-201.
- Banfield C, Gupta S, Marino M, Lim J, Affrime M: O sumo de toranja reduz a biodisponibilidade oral da fexofenadina mas não da desloratadina. Clin Pharmacokinet 2002; 41: 311-318.
- Hondeghem LM, Dujardin K, Hoffmann P, Dumotier B, De Clerck F: O prolongamento QTc induzido por drogas subestima perigosamente o potencial proarrítmico: lições de terfenadina. J Cardiovasc Pharmacol 2011; 57: 589-597.
- Compalati E, Baena-Cagnani R, Penagos M, Badellino H, Braido F, Gómez RM, Canonica GW, Baena-Cagnani CE: Revisão sistemática sobre a eficácia da fexofenadina na rinite alérgica sazonal: uma meta-análise de ensaios clínicos aleatórios, duplo-cegos e controlados por placebo. Int Arch Allergy Immunol 2011; 156: 1-15.
PRÁTICA DO GP 2013; 8(3): 14-17









