As leucemias infantis são tratadas de uma forma adaptada ao risco e são curáveis na maioria dos casos. Na leucemia linfoblástica aguda, um dos factores prognósticos mais importantes – para além dos marcadores prognósticos clássicos como o subtipo leucémico e as alterações citogenéticas nas explosões leucémicas – é a determinação da doença residual mínima após a indução da terapia. Os desenvolvimentos actuais visam um tratamento mais eficaz dos subtipos de leucemia anteriormente resistentes, bem como uma redução da toxicidade da terapia.
As leucemias são os cancros da infância e adolescência mais comuns nos países industrializados, representando cerca de 35% de todas as neoplasias. Na Suíça, diagnosticamos todos os anos 55-65 novos casos. A incidência actual em crianças com menos de 15 anos é de cerca de 4,9 casos/100.000 habitantes. As duas formas mais importantes de leucemia infantil são a leucemia linfoblástica aguda (ALL) (aprox. 82%) e a leucemia mielóide aguda (AML) (aprox. 15%). Apenas raramente ocorrem síndromes mielodisplásticas e leucemias mielóides crónicas em crianças.
Leucemia linfática aguda
A infância mais comum de TODOS é a B-precursor ALL, que se desenvolve a partir de células imaturas da série B do sistema linfático. TODOS os T-lymphoiesis ocorrem com menos frequência. Uma forma especial é a leucemia madura da célula B, que se baseia numa transformação maligna da célula B madura e é entendida como uma manifestação leucémica do linfoma de Burkitt. A causa do desenvolvimento da leucemia é ainda pouco clara e continua a ser o foco de muitos estudos epidemiológicos. Os factores conhecidos mas que raramente ocorrem são a radiação ionizante e as síndromes congénitas. No entanto, isto explica menos de 10% de todas as doenças. Estima-se que cerca de 1% das crianças nascidas com síndrome de Down desenvolvem leucemia (ALL ou AML) durante os primeiros cinco anos. O seu risco é cerca de 20 vezes maior em comparação com crianças saudáveis. Contudo, a mieloproliferação transitória ocorre ainda mais frequentemente (em 3-10%) nestas crianças em idade neonatal, o que pode ocasionalmente transformar-se mais tarde em leucemia. Outras alterações congénitas mais raras com risco aumentado de leucemia são a ataxia teleangiectásica, a síndrome de Fanconi e outras síndromes associadas a uma imunodeficiência ou aumento da fragilidade cromossómica.
Várias observações conduziram a diferentes hipóteses relativamente a um desenvolvimento de leucemia associado à infecção: por exemplo, o facto de que TODOS ocorrem mais frequentemente entre o segundo e o quinto ano de vida, que uma acumulação da doença pode ser vista em países industrializados, ou que no passado a agregação ocorreu repetidamente especialmente em regiões de novas aglomerações. [1,2].
Sintomas de TODOS
Em primeiro plano estão sintomas devidos à supressão da formação normal de células sanguíneas na medula óssea, tais como palidez e fadiga devido a anemia ou tendências a sangramento devido a trombocitopenia. As infiltrações conduzem frequentemente a dores ósseas difusas e artropatias alternadas, que ocasionalmente se manifestam em crianças pequenas como relutância em mover-se ou mesmo recusa em andar. Além disso, pode ocorrer inchaço dos gânglios linfáticos e organomegalia.
Diagnóstico de TODOS
No sangue, são frequentemente encontradas alterações em pelo menos duas séries de células sanguíneas, mais frequentemente trombocitopenia com anemia simultânea. A contagem de leucócitos pode ser normal, diminuída ou aumentada. A morfologia do hemograma pode fornecer pistas de diagnóstico importantes; o diagnóstico final é feito por meio de uma aspiração de medula óssea. Além de examinar a morfologia, o imunofenótipo das explosões leucémicas é determinado por meio da citometria de fluxo (FACS) e é realizada uma análise cromossómica. A imunofenotipagem permite determinar a fase de desenvolvimento do clone celular correspondente.
O subtipo mais comum de leucemia nas crianças, o chamado “TODO comum”, caracteriza-se pela expressão dos marcadores de células B CD10 e CD 19. Uma expressão de antigénios mielóides, que normalmente não é prognosticada, pode ser detectada em quase metade de TODOS os casos.
Actualmente, o diagnóstico citogenético e genético molecular desempenha um papel importante. É importante reconhecer os subgrupos mais importantes, uma vez que têm implicações terapêuticas. Por um lado, procuram-se alterações cromossómicas numéricas tais como hiper ou hipodiploidia, bem como alterações estruturais tais como translocações, por exemplo t(12;21) (fusão dos genes ETV6/RUNX1) ou t(9;22) (fusão de BCR/ABL1), rearranjos MLL (MLL 11q23) e outras alterações. Classicamente, estas alterações são detectadas por meio de citogenética convencional (banda G) e/ou hibridação fluorescente in situ (FISH) nas células de leucemia. As alterações cromossómicas mais comuns em TODOS são mostradas no Quadro 1 .
A resposta à terapia é um parâmetro prognóstico muito importante. Nos últimos anos, a medição da doença residual mínima (DRM) da medula óssea foi estabelecida como parte dos diagnósticos de acompanhamento para avaliar a resposta à terapia. Essencialmente, são utilizados dois métodos que se complementam na prática clínica diária. O método mais sensível é o controlo dos rearranjos dos receptores de imunoglobulina e de células T. Inicialmente, procuram-se rearranjos clonais específicos da leucemia, que são seguidos por meio de PCR quantitativa em pontos de tempo terapêuticos específicos. O limite de detecção assim alcançado é aproximadamente uma célula de leucemia por 100.000 células normais da medula óssea. Uma técnica para a medição do MRD que é cerca de um nível de registo menos sensível baseia-se na monitorização do imunofenótipo associado à leucemia pela FACS. Uma sensibilidade de 0,001% pode ser alcançada [3].
Terapia e prognóstico de TODOS
A adaptação contínua do tratamento para TODOS ao longo dos últimos 40 anos levou a um aumento dramático da probabilidade de sobrevivência. O Grupo de Estudos ALL-BFM, uma associação de centros de oncologia pediátrica alemã, austríaca e suíça, tem contribuído significativamente para esta melhoria em numerosos estudos terapêuticos aleatórios de grande escala desde 1976.
A terapia de indução visa alcançar a remissão e restauração da hematopoiese normal dentro das primeiras quatro a seis semanas de terapia. Isto é possível com as modalidades actuais em cerca de 98% de todos os pacientes com as três drogas cortisona, vincristina e asparaginase; em alguns grupos de estudo, os pacientes recebem também uma anthracycline [4–6]. Nos protocolos ALL-BFM que usamos, a indução da remissão é iniciada por sete dias de monoterapia com prednisona e administração intratérmica de metotrexato. Desta forma, as complicações devidas à decomposição celular podem ser evitadas na maioria dos casos, e uma avaliação inicial da sensibilidade das células de leucemia a estes medicamentos já pode ser investigada (resposta de prednisona). A consolidação (metotrexato de alta dose), que tem um efeito particularmente forte no SNC, é seguida de terapia de reintrodução; foi demonstrado que o uso repetido de terapia de indução após um período de consolidação pode reduzir significativamente o risco de recidiva [5,7]. Componentes igualmente indispensáveis para o tratamento bem sucedido da leucemia são a terapia extra-compartimental (tratamento preventivo do SNC) e a terapia de manutenção contínua prolongada, que permite a remissão através da supressão de clones resistentes à leucemia. Hoje em dia, a prevenção da recorrência do SNC é feita principalmente com drogas, por um lado através de injecções de metotrexato intratecal, e por outro lado através de drogas citostáticas de acção sistémica que se infiltram no cérebro (por exemplo, metotrexato de alta dose). Isto tornou possível limitar a radioterapia precoce do SNC a situações de risco muito especiais; embora isto leve a uma redução dramática das recorrências do SNC, está associado a efeitos tardios não negligenciáveis [4,5,8].
De particular relevância hoje em dia são as estratégias terapêuticas adaptadas ao risco, que resultam de vários parâmetros biológicos tais como a citogenética e a resposta terapêutica inicial (Tab. 1). Por um lado, o objectivo é melhorar o prognóstico das leucemias de alto risco através da intensificação da terapia ou do uso selectivo de novas substâncias; por outro lado, o objectivo é evitar toxicidade desnecessária com um prognóstico favorável. A contagem de células, idade, sexo, citogenética e a resposta inicial dos linfoblastos no sangue periférico à administração de prednisona nos primeiros oito dias são factores que influenciam o prognóstico [9]. A resposta adicional no contexto da indução terapêutica é também um parâmetro prognóstico relevante.
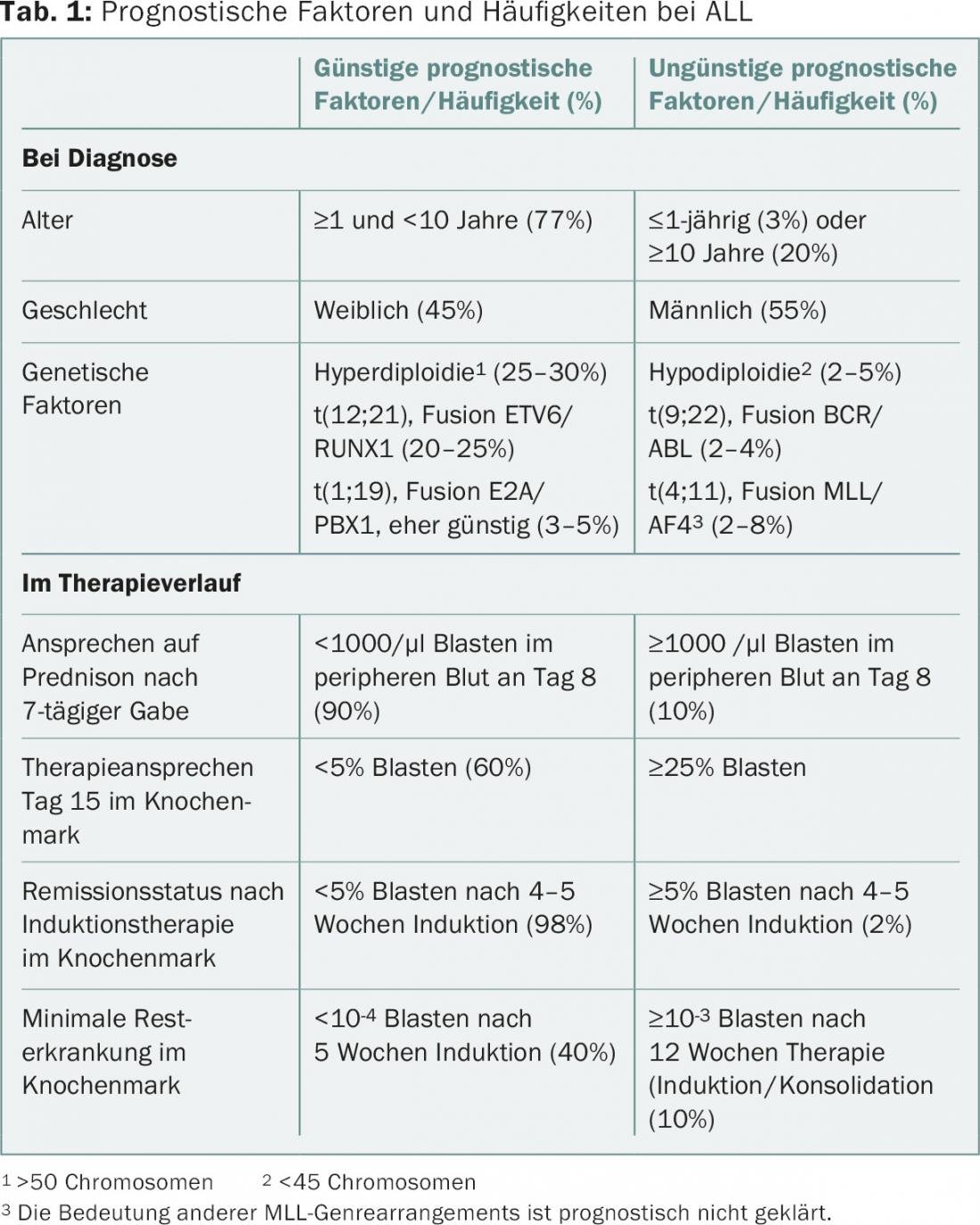
Estudo terapêutico AIEOP-BFM ALL 2009
A maioria dos centros na Suíça tratam crianças com TODOS no estudo multicêntrico AIEOP-BFM ALL 2009. Para além dos “antigos” países BFM Suíça, Alemanha e Áustria, quatro outros países (Itália, Israel, Austrália, República Checa) acordaram um protocolo de tratamento comum (Fig. 1) . O objectivo do estudo é incluir em conjunto cerca de 4000 crianças e adolescentes com TODAS e testar aleatoriamente várias questões relevantes para a terapia. A atribuição aos três grupos de risco SR (risco padrão), MR (risco médio) e HR (risco elevado) baseia-se, por um lado, em critérios biológicos como a hipodiploidia ou rearranjo MLL, e, por outro lado, na avaliação da resposta terapêutica utilizando as várias técnicas de MRD. Os seguintes factores estão incluídos na estratificação do risco: a resposta da leucemia à pré-estágio de 7 dias com esteróides, a diminuição das células de leucemia na medula óssea medida pela FACS no dia 15 da terapia e a doença residual mínima nos dias 33 e 78 após o início da terapia, medida pela diminuição dos rearranjos imunitários e receptores de células T específicos da leucemia com PCR quantitativa. [10].
Em crianças com uma resposta muito boa aos primeiros 15 dias de terapia, medida pelo FACS-MRD, será testada uma redução das doses de antraciclina durante a fase de indução para duas em vez de quatro doses (R1). Para o maior grupo de doentes MR, o benefício – no que respeita ao risco de recorrência – de nove doses adicionais de uma asparaginase peguilada de acção prolongada durante 20 semanas no final da fase de quimioterapia intensiva será testado aleatoriamente (R2). A terceira aleatorização diz respeito aos doentes de RH que, apesar dos esforços intensivos das últimas décadas, ainda só atingem uma taxa livre de recorrência de cerca de 50%. Neste grupo de pacientes, a terapia já deveria ser intensificada na fase de indução com quatro administrações adicionais de asparaginase (RHR).
Tratamento de recidiva
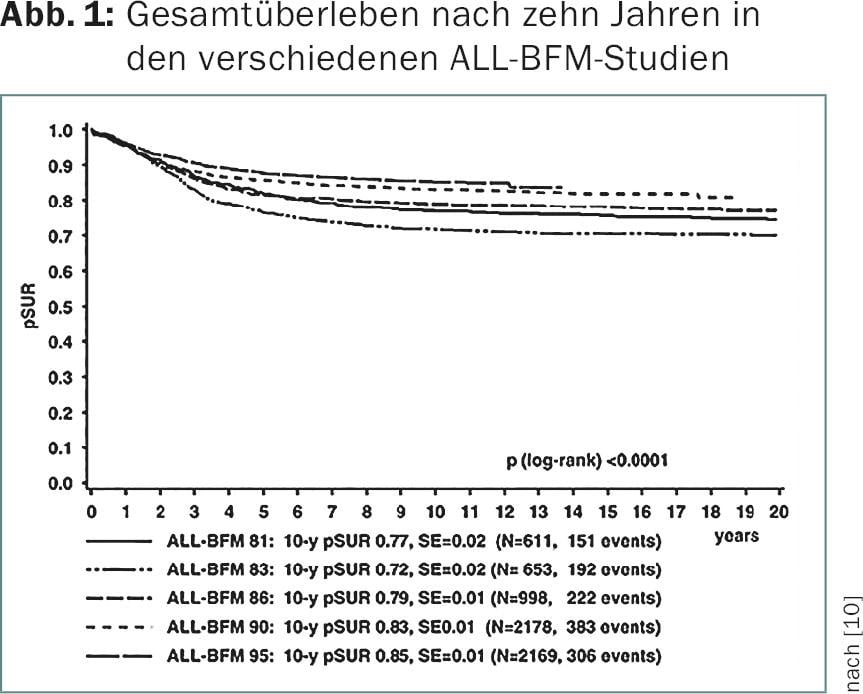
Os resultados da terapia primária bem como os protocolos de recaída das leucemias têm melhorado ao longo do tempo. Com as actuais terapias adaptadas ao risco, são alcançadas taxas de sobrevivência a longo prazo de aproximadamente 85% (Fig. 2) [7]. Isto também levou a um ajustamento contínuo da indicação para terapias de altas doses com reinfusão de células estaminais. No âmbito de um protocolo internacional de transplante de células estaminais (ALL-SCT-BFM 2003), foi feita uma tentativa de estabelecer definições claras para a indicação de transplante alogénico de células estaminais, tanto no contexto da terapia primária como no caso de recaídas. Actualmente, existe uma indicação para o transplante de medula óssea como parte da terapia primária para certos subgrupos citogenéticos prognósticos desfavoráveis, tais como o t(9;22), ou para uma resposta insuficiente à terapia de indução [11].
A experiência do grupo BFM mostrou que o sucesso do tratamento das recaídas depende do tempo da recaída, do padrão da leucemia e do subtipo da leucemia [12]. A resposta terapêutica após a indução terapêutica renovada e, portanto, a dinâmica do declínio da doença residual mínima é também aqui de particular importância prognóstica; em conformidade, outros elementos terapêuticos, tais como um transplante de medula óssea, são alinhados em conformidade [13]. Há uma esperança justificada de que a terapia orientada com anticorpos ou inibidores de cinase mais específicos seja possível no futuro para vários subtipos de TODOS. Um exemplo é o anticorpo CD19 (blinatumomab), que tem sido capaz de induzir taxas de remissão encorajadoras em TODOS os ensaios das fases I e II [14]. Outras abordagens terapêuticas possíveis de terapias específicas envolvem interacções com vias de sinalização dentro das células com leucemia. Isto inclui, por exemplo, a supressão da expressão do gene CRLF2 ou a activação anormal do gene JAK.
Leucemia mielóide aguda
Em AML, um caule mielóide ou célula progenitora é afectado, o que é responsável pela formação de granulócitos, monócitos, plaquetas ou eritrócitos. Ao contrário de TODOS, a subtilografia imunológica é menos importante em AML. Também em AML, além do subtipo morfológico (M0-M7 segundo a classificação FAB), os factores genéticos, bem como a resposta à terapia e a monitorização da doença residual, são parâmetros importantes na avaliação prognóstica [15]. Traduções como t(8;21) especialmente em AML, FAB M1/M2, t(15;17) típico de AML, FAB M3, inversão(16) em AML e FAB M4 Eo são factores favoráveis, enquanto certos subtipos com rearranjo do gene MLL (11q23) ou mutação do gene FLT3 são prognosticalmente desfavoráveis (Tabela 2) [16].

Também em AML parece não haver uma terapia de tamanho único para todos os subgrupos. Enquanto que na leucemia promielocítica aguda (AML, FAB M3) a introdução de ácido retinóico reduziu significativamente a taxa de complicações e melhorou a taxa de cura, outros subtipos são quase impossíveis de controlar sem o transplante de medula óssea. A hiperleucocitose em AML bem como o subtipo FAB M3 é uma situação de emergência e requer uma intervenção rápida uma vez que o risco de hemorragia é significativamente aumentado.
Os medicamentos cruciais para a indução da remissão em AML são a citarabina e as anthraciclinas [17,18]. O etoposídio também é frequentemente adicionado. O benefício adicional da quimioterapia pós-indução, mais uma vez com citarabina de alta dose, foi demonstrado várias vezes. Nos últimos anos, a 2-CDA (2-cloro-2-deoxiadenosina) também tem sido utilizada com sucesso nas leucemias de alto risco, especialmente em combinação com a citarabina. Esta combinação leva a uma inibição máxima da síntese de ADN. Em contraste com TODOS, a utilização da terapia de manutenção ainda é controversa em AML [19]. A abordagem à terapia orientada para o SNC é também inconsistente. Vai desde a monoterapia intratecal com citarabina ou metotrexato até à terapia tripla com citarabina, metotrexato e hidrocortisona, com ou sem radioterapia adicional. No entanto, em geral, a radioterapia tem sido significativamente reduzida ou mesmo completamente omitida em AML nos últimos anos.
A taxa de cura para AML depende fortemente dos parâmetros mencionados, mas hoje atinge até 70%. O transplante alogénico de células estaminais ainda é generosamente recomendado em certos grupos de estudo se estiver disponível um dador irmão compatível. Outros preferem uma indicação adaptada ao risco para o transplante de células estaminais (SCT). No entanto, em geral, há uma relutância crescente em recomendar a SCT. Em qualquer caso, a SCT é indicada em caso de remissão incompleta ou de recorrência. A esperança está ligada à utilização orientada de inibidores de cinase para as mutações detectadas, tais como as mutações FLT3 ou KIT.
Efeitos tardios dos tratamentos contra a leucemia
Com o aumento das taxas de sobrevivência após a leucemia, a atenção aos efeitos tardios aumenta naturalmente. Por exemplo, a cardiotoxicidade a longo prazo após terapia para LMA deve ser considerada. A osteonecrose e outros eventos vasculares, que são comuns em TODOS, também podem causar morbidades a longo prazo. Após a TCT, os efeitos tardios são mais frequentes e mais relevantes. O risco de malignidades secundárias depende novamente dos fármacos citostáticos utilizados, e há também um risco acrescido após a radioterapia.
Literatura:
- Kinlen L: Infecções e factores imunitários no cancro: o papel da epidemiologia. Oncogene 2004; 23: 60-75.
- Greaves M: Infecção, respostas imunitárias e a etiologia da leucemia infantil. Nat Rev Cancer 2006; 6(3): 193-203.
- Campano D: Monitorização mínima de doenças residuais na leucemia linfoblástica aguda infantil. Curr Opinião Hematol 2012; 19: 313-318.
- Pui CH, Evans WE: Tratamento da leucemia linfoblástica aguda infantil. N Engl J Med 2006; 354: 166-178.
- Möricke A, et al.: A terapia ajustada ao risco de leucemia linfoblástica aguda pode diminuir a carga do tratamento e melhorar a sobrevivência: resultados do tratamento de 2169 pacientes pediátricos e adolescentes não seleccionados inscritos no ensaio ALL-BFM 95. Sangue 2008; 111(9): 4477-4489.
- Schrappe M, et al: Resultados a longo prazo de quatro ensaios consecutivos na infância TODOS realizados pelo grupo de estudo ALL-BFM de 1981 a 1995. Berlin-Frankfurt-Münster. Leucemia 2000; 14(12): 2205-2222.
- Möricke A, et al: Resultados a longo prazo de cinco ensaios consecutivos em leucemia linfoblástica aguda infantil realizados pelo grupo de estudo ALL-BFM de 1981 a 2000. Leucemia 2010; 24(2): 265-284.
- Kamps WA, et al: tratamento orientado para BFM para crianças com leucemia linfoblástica aguda sem irradiação craniana e redução do tratamento para doentes de risco padrão: resultados do protocolo DCLSG ALL-8 (1991-1996). Leucemia 2002; 16(6): 1099-1111.
- Schrappe M, et al: Melhor resultado na leucemia linfoblástica aguda infantil apesar da redução do uso de antraciclinas e radioterapia craniana: resultados do ensaio ALL-BFM 90. Grupo de Estudo Alemão-Austríco-Suiço ALL-BFM. Sangue 2000; 95(11): 3310-3322.
- Protocolo Internacional de Tratamento Colaborativo para Crianças e Adolescentes com Leucemia Linfoblástica Aguda. http://clinicaltrials.gov/show/NCT01117441.
- Balduzzi A, et al: Quimioterapia versus transplante alogénico para a leucemia linfoblástica aguda infantil de muito alto risco na primeira remissão completa: comparação por randomização genética num estudo prospectivo internacional. Lancet 2005; 366: 635-642.
- Tallen G, et al: Resultado a longo prazo em crianças com recidiva de leucemia linfoblástica aguda após estratificação do ponto temporal e do local do colapso e intensificação da quimioterapia multi-droga de curta duração: resultados do ensaio ALL-REZ BFM 90. J ClinOncol 2010; 28: 2339-2347.
- Eckert C, et al: Minimal residual disease after induction is the strongest predictor of prognosis in intermediate risk recapsed lymphoblastic leukaemia aguda – Long-term results of trial ALL-REZ BFM P95/96. Eur J Cancer 2012; http://dx.doi.org/10.1016/j.ejca.2012.11.010.
- Schlegel P, et al.: A leucemia linfoblástica aguda póstransplante pediátrica recaída/refractária B-precursor mostra uma remissão duradoura por terapia com a célula T envolvente cegumomab anticorpo bi-específico. Haematologica 2014; 99(7); 1212-1219.
- Abrahamsson J, et al: Terapia de indução guiada por resposta em leucemia mielóide aguda pediátrica com excelente taxa de remissão. J ClinOncol 2011; 29(3): 310-315.
- Creutzig U, et al.: Comité AML do Grupo Internacional de Estudos BFM. Diagnóstico e gestão da leucemia mielóide aguda em crianças e adolescentes: recomendações de um painel internacional de peritos. Sangue 2012; 120(16): 3187-3205.
- Creutzig U, et al: Estratégias de tratamento e resultados a longo prazo em doentes pediátricos tratados em quatro ensaios consecutivos de AML-BFM. Leucemia 2005; 19(12): 2030-2042.
- Creutzig U, et al: Menos toxicidade pela optimização da quimioterapia, mas não pela adição do factor estimulante da colónia de granulócitos em crianças e adolescentes com leucemia mielóide aguda: resultados de AML-BFM 98. J ClinOncol 2006; 24(27): 4499-4506.
- Perel Y, et al.: Grupo LAME da Sociedade Francesa de Pediatria-Hematologia e Imunologia. Impact of addition of maintenance therapy to intensive induction and consolidation chemotherapy for childhood acute myeloblasticleukemia: results of a prospective randomized trial, LAME 89/91. J Clin Oncol 2002; 20(12): 2774-2782.
InFo ONCOLOGY & HEMATOLOGY 2015; 14(5): 8-12