O tratamento profilático com concentrados de factor é agora padrão no tratamento de pacientes com hemofilia grave A. Os concentrados de factor VIII e de factor IX têm uma meia-vida relativamente curta e, consequentemente, têm frequentemente de ser aplicados por via intravenosa. Há já alguns meses, tanto os produtos de factor VIII como os produtos de factor IX com meia-vida prolongada têm sido aprovados na Suíça. Várias opções terapêuticas alternativas para o tratamento da hemofilia poderiam atingir a maturidade do mercado nos próximos anos.
Embora em princípio todas as perturbações da coagulação congénita que levam a uma tendência crescente para a hemorragia possam ser subsumidas sob o termo “hemofilia”, num sentido mais restrito descreve duas perturbações definidas da coagulação do sangue: por um lado, a deficiência do factor VIII de coagulação do sangue (hemofilia A) e, por outro lado, a deficiência do factor IX de coagulação do sangue (hemofilia B). Ambas as perturbações são herdadas de uma forma recessiva ligada ao X. Para além da hemofilia hereditária, existem também as muito raras formas adquiridas (hemofilia auto-imune), que não serão aqui discutidas.
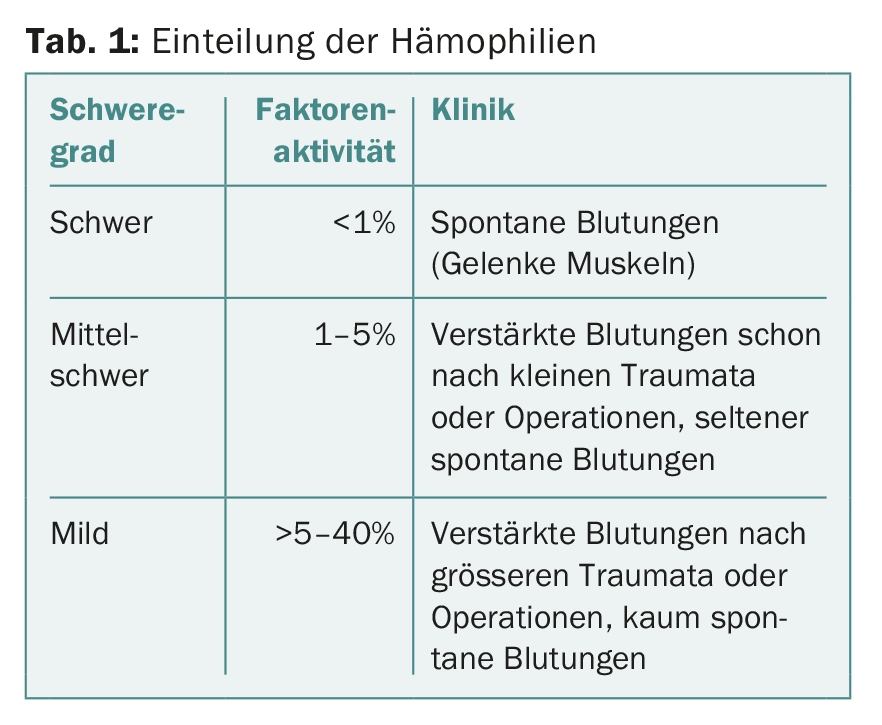
Nas hemofilias hereditárias, a actividade residual do factor VIII ou IX correlaciona-se relativamente bem com a apresentação clínica. Isto torna possível classificar a hemofilia A e B de acordo com a actividade residual do factor VIII e IX respectivamente (tab. 1).
Tratamento da hemofilia
A invenção do tratamento de substituição intravenosa com concentrados de factores do plasma humano há mais de 50 anos reduziu significativamente a mortalidade e morbilidade da hemofilia, reduzindo os episódios de hemorragia. Contudo, esta história de sucesso foi gravemente manchada pela transmissão de doenças virais como o VIH e a hepatite C nos anos 70 e 80. No entanto, graças à implementação de melhores métodos de rastreio e inactivação de vírus, bem como ao desenvolvimento de preparações de factores recombinantes, a segurança das preparações foi maciçamente melhorada, de modo a que não foram relatadas mais transmissões de vírus desde 1990. O principal risco de tratamento com concentrados de factores actualmente é a ocorrência de inibidores, que são observados em cerca de 20-30% dos doentes com hemofilia A grave (um pouco menos frequentemente em hemofilia B). Como o nome sugere, estes inibidores inibem os factores de coagulação fornecidos e tornam o tratamento de substituição ineficaz. Em muitos casos, no entanto, é possível livrar-se dos inibidores por meio de terapias de imunotolerância complexas.
Nos doentes com hemofilia grave, a chamada “profilaxia” é padrão. Os concentrados de factores são (geralmente) administrados por via intravenosa duas a três vezes por semana, de tal forma que a hemorragia espontânea (articular) é largamente evitada aumentando as concentrações de plasma do factor de coagulação em causa. A frequência de injecção depende em última análise da meia-vida do factor de coagulação em causa (factor VIII aprox. 12 h, factor IX aprox. 18 h) [1].
As injecções intravenosas relativamente frequentes são experimentadas por muitos pacientes como aborrecidas, e no final não se consegue, no entanto, uma normalização contínua do sistema de coagulação.
Novos preparativos de factores
Existe algum potencial de melhoria no desenvolvimento de novas opções de tratamento para a hemofilia: meia-vida mais longa (portanto menos injecções e melhor protecção), formas alternativas de administração (oral ou subcutânea em vez de intravenosa), menor imunogenicidade (menos formação de corpos inibidores) e, em princípio, melhor eficácia.
Certamente as mais avançadas são as técnicas para prolongar a meia-vida. Basicamente, os factores de coagulação recombinantes são modificados de tal forma que se degradam menos rapidamente. Várias destas preparações de “longa duração” receberam autorização de comercialização na Suíça nos últimos meses. As abordagens utilizadas são, por um lado, a modificação química dos factores (acoplamento a polietilenoglicóis (PEGylation)), fusão com outras proteínas (região Fc da imunoglobulina G, albumina recombinante) ou modificação da sequência proteica direccionada. Por uma questão de exaustividade, deve mencionar-se aqui que algumas preparações clássicas recombinantes do factor VIII também entraram no mercado nos últimos anos, que tendem a ter mais meia-vida do que as suas antecessoras graças a processos de fabrico optimizados (por exemplo, glicolização optimizada). No entanto, estas preparações não são consideradas “meia-vida prolongada” no sentido estrito.
Em seguida, serão brevemente apresentados os métodos utilizados para alcançar uma meia-vida prolongada.
PEGylation: Nesta técnica, uma a várias 20-60 kDa As moléculas de PEG estão ligadas a uma molécula de factor VIII ou IX. Os diferentes fabricantes utilizam conceitos diferentes. É essencial que as moléculas de PEG estejam ligadas à molécula de factor de tal forma que o seu efeito hemostaseológico não seja prejudicado.
Devido ao revestimento com moléculas de PEG, as moléculas de factor são menos acessíveis às proteases; além disso, o PEGylation causa um aumento do tamanho, bem como uma melhor solubilidade da água. Tudo isto leva a uma semi-vida prolongada dos factores PEGylated [2].
Tecnologia de proteína de fusão: Ao fundir uma proteína de coagulação com outra proteína que tem naturalmente uma meia-vida significativamente mais longa na corrente sanguínea, a própria meia-vida da proteína de coagulação pode, em última análise, ser aumentada.
Especificamente, a região Fc da imunoglobulina G (IgG-Fc) ou albumina recombinante é utilizada para a fusão. Tanto a IgG-Fc como a albumina têm meia-vida muito longa porque estão protegidas da degradação lisossómica através de uma espécie de mecanismo de reciclagem [3].
Modificação da sequência específica do local: Nesta técnica, a molécula de factor VIII é modificada de modo a ter uma afinidade de ligação significativamente maior com a sua molécula de factor von Willebrand portadora. Com cerca de 15 horas, o factor von Willebrand tem uma meia-vida ligeiramente mais longa do que o factor VIII. Devido à forte ligação da molécula do factor VIII à molécula do factor von Willebrand, a meia-vida do factor VIII aproxima-se da do factor von Willebrand [4].
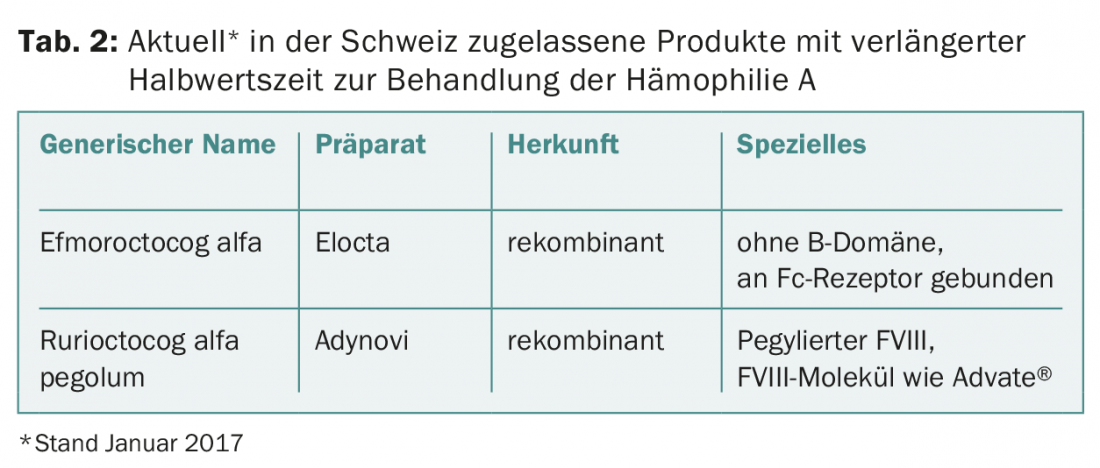
Prorrogação da meia-vida dos produtos FVIII: A meia-vida do factor VIII é finalmente limitada pelo factor von Willebrand [5]. Isto porque o factor von Willebrand, como proteína transportadora, protege o factor VIII da degradação proteolítica. Todas as medidas acima descritas permitem, portanto, prolongar a meia-vida do factor VIII (cerca de 12 h) no máximo por cerca de 1,5 vezes. Embora isto não seja muito, significa já um progresso clinicamente mensurável. Por exemplo, se um paciente precisar de três injecções por semana com um produto convencional, poderia alcançar os mesmos níveis de vale com apenas duas injecções por semana com um produto de meia-vida prolongada. Por outro lado, se o mesmo paciente mantivesse a sua frequência de injecção de três vezes por semana com o novo produto, poderia atingir níveis de vale significativamente mais elevados. Isto faria sentido, por exemplo, se ele ainda tivesse tido uma hemorragia de ruptura apesar da profilaxia regular [6]. Os produtos do factor VIII de meia-vida actualmente aprovados na Suíça estão listados no quadro 2 .
Prorrogação da meia-vida dos produtos do factor IX: O factor IX de coagulação normal do sangue tem uma meia-vida de cerca de 18 a 20 horas. Ao contrário do factor VIII, o factor IX não está ligado a uma proteína transportadora. As medidas descritas acima têm, portanto, um efeito muito mais impressionante no factor IX do que no factor VIII: tanto a PEGylation como as proteínas de fusão causam um prolongamento da meia-vida por um factor 5, ou seja, para cerca de 90 horas.
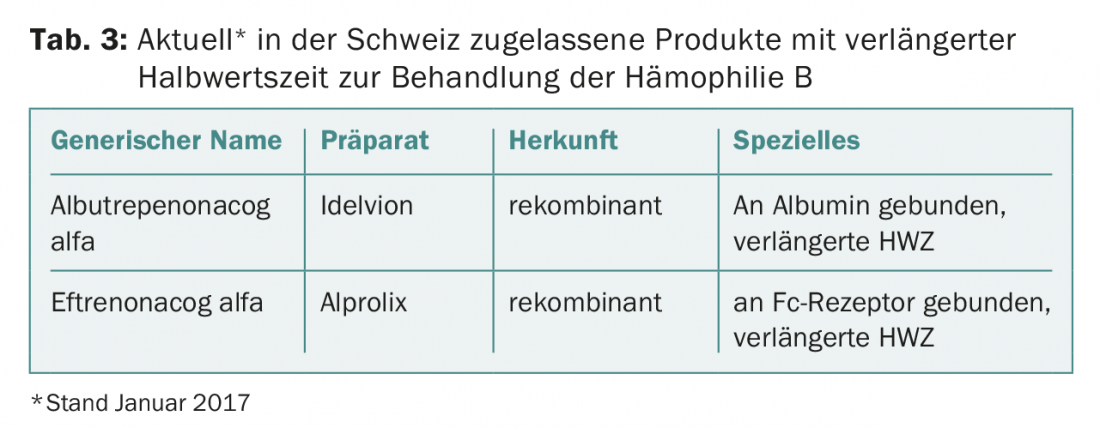
Este resultado impressionante abre novas estratégias terapêuticas. As preparações de factor IX com meia-vida prolongada só precisam de ser aplicadas de uma a duas semanas para a profilaxia (preparações normais de factor IX aproximadamente duas vezes por semana) e ainda podem ser atingidos níveis mais elevados nos vales [7]. Os produtos do factor IX de meia-vida actualmente aprovados na Suíça estão listados no quadro 3.
Abordagens terapêuticas alternativas
Para além dos métodos acima mencionados, que em última análise todos visam optimizar a terapia de substituição com concentrados de factores, numerosas abordagens terapêuticas alternativas estão agora também a ser seguidas. Neste caso, é feita uma tentativa de conseguir uma activação de coagulação suficiente, apesar da hemofilia existente, através de manipulações direccionadas do sistema de coagulação a fim de, pelo menos, evitar hemorragias espontâneas. Possíveis vantagens de se desviar do conceito clássico de substituição de factores incluem evitar o problema do desenvolvimento de inibidores e a possibilidade de formas alternativas de administração.
Uma destas abordagens alternativas consiste em bloquear a anticoagulação fisiológica. Especificamente, está a ser realizada investigação para inibir especificamente ou desregulamentar o Inibidor da Via Patológica do Factor Tecidual (TFPI) ou antitrombina. O Fitusiran, por exemplo, é uma molécula que pode visar o antitrombina. Esta molécula de interferência de RNA, que pode ser aplicada subcutaneamente, inibe especificamente a formação de antitrombina. Isto leva a uma melhor geração de trombina no sistema de coagulação, resultando num melhor controlo da hemorragia no doente com hemofilia [8]. A substância Fitusiran já foi testada clinicamente com sucesso (fase 1/2).
Outra substância interessante que está a ser submetida com sucesso a um programa de ensaios clínicos até agora é o emicizumab de anticorpos biespecíficos. Este anticorpo imita até certo ponto a função do factor VIII activado ligando-se simultaneamente aos factores de coagulação IX e X, resultando numa maior activação do factor X e acelerando assim a coagulação do sangue. O emicizumabe pode ser injectado subcutaneamente e é também eficaz quando estão presentes inibidores contra o factor VIII. No entanto, o anticorpo não atinge a mesma eficiência biológica que o factor de coagulação natural VIII [9].
Outra abordagem terapêutica que ainda está a ser seguida é a terapia genética. Em estudos, é possível melhorar a actividade endógena do factor IX a longo prazo em doentes com hemofilia (grave) B, mas a normalização da actividade do factor IX ainda não foi alcançada. Na hemofilia A, a terapia genética ainda não é tecnicamente possível [9].
Conclusão
Os muitos novos produtos que entraram no mercado nos últimos meses e que entrarão no mercado nos próximos anos são promissores. Algumas das preparações mencionadas têm definitivamente o potencial de revolucionar o tratamento da hemofilia. Contudo, ainda não é claro se, na melhor das hipóteses, os custos potencialmente elevados, especialmente das abordagens terapêuticas alternativas discutidas acima, poderiam influenciar a utilização clínica.
Precisamente devido às numerosas opções de que dispomos nos países desenvolvidos para o tratamento da hemofilia, não devemos esquecer que a nível mundial muitos destes doentes ainda não têm acesso suficiente aos concentrados de factores.
Literatura:
- Ljung R: Aspectos do tratamento profiláctico da hemofilia. Diário de trombose. 2016;14(Suplemento 1): 30.
- Ivens IA, et al: PEGylated therapeutic proteins for haemophilia treatment: a review for haemophilia careegivers. Hemofilia: o jornal oficial da Federação Mundial de Hemofilia. 2013;19(1): 11-20.
- Powell JS: Concentrados de factor de coagulação de acção mais longa para hemofilia. Diário de trombose e hemostasia: JTH. 2015;13 Suppl 1: S167-75.
- Comprar Y, Liu T, et al.: Uma única variante em cadeia da proteína de fusão Fc factor VIII retém a eficácia normal in vivo mas apresenta uma actividade in vitro alterada. PloS um. 2014;9(11): e113600.
- Pipe SW, et al: Life in the shadow of a dominant partner: a associação FVIII-VWF e as suas implicações clínicas para a hemofilia A. Blood. 2016.
- Tiede A: Factor VIII de meia-vida prolongado para o tratamento da hemofilia A. Diário de trombose e hemostasia: JTH. 2015;13 Suppl 1: S176-9.
- Nazeef M, Sheehan JP: Novos desenvolvimentos na gestão da hemofilia moderada a séria B. Journal of blood medicine. 2016;7: 27-38.
- Sehgal A, et al.: Uma terapêutica de RNAi visando o antitrombina para reequilibrar o sistema de coagulação e promover a hemostasia em hemofilia. Medicina natural. 2015;21(5): 492-7.
- Shima M, Hanabusa H, Taki M, et al: Factor VIII-Mimetic Function of Humanized Bispecific Antibody in Hemophilia A. The New England journal of medicine. 2016;374(21): 2044-53.
InFo ONCOLOGy & HEMATOLOGy 2017; 5(1): 5-9









