
Se se suspeitar de um tumor secante de catecolaminas, a determinação de metanefrinas no plasma livre é um método de diagnóstico sensível. A hipertensão paroxística é um sintoma comum. Dependendo dos resultados laboratoriais, deve ser realizado um exame de imagem para localizar o tumor. No caso de tumores não renováveis ou metastáticos, existem novas terapias experimentais baseadas na assinatura genética e molecular individual, nas quais se deposita muita esperança. Neste contexto, as análises genéticas estão a ganhar importância.
Os feocromocitomas (PCCs) são tumores produtores de catecolaminas que se originam das células cromatográficas da medula adrenal. Os Paragangliomas (PGL) são tumores produtores de noradrenalina e dopamina do paraganglia simpático e parassimpático [1]. O quadro clínico e o procedimento de diagnóstico inicial são muito semelhantes em ambos os tipos de tumores secadores de catecolaminas [2]. Um sintoma típico do feocromocitoma é a hipertensão, que ocorre paroxisticamente em cerca de metade dos doentes afectados. Outros sinais da doença incluem transpiração, palpitações, dores de cabeça, tremores ou náuseas. Na maioria dos casos, os PGL são facilmente curáveis por ressecção completa, mas existem diferenças significativas no risco de malignidade dependendo do genótipo e da localização do tumor. Os tumores que metastasisam são considerados malignos, ou seja, cerca de 10-15% de todos os PCC e 35-40% de todos os PGL [1]. Todos os doentes com feocromocitomas/paragangliomas (PPGL) devem ser submetidos a testes genéticos, salientou a Prof. Dra. med. Svenja Nölting, médica sénior da Clínica de Endocrinologia, Diabetologia e Nutrição Clínica do Hospital Universitário de Zurique, na reunião anual da Sociedade Suíça de Endocrinologia e Diabetologia (SGED) em Novembro passado [1].

Tipagem em grupo: assinatura genética e molecular
“Cerca de 70% de todos os PPGLs podem ser atribuídos a um dos três grupos moleculares com base na sua mutação subjacente”, explicou o orador [1,3]. A maioria dos tumores pertence aos aglomerados 1 e 2; pouco se sabe até agora sobre o aglomerado 3. Os tumores do aglomerado 1 baseiam-se na activação de vias de sinalização de pseudo-hipoxia, os tumores do aglomerado 2 na activação de vias de sinalização dependentes da tirosina [4]. Em 30-35% dos casos há mutações autossómicas herdadas da linha germinal, 35-40% são devidas a mutações somáticas. Os tumores de aglomerado 1 são mais frequentemente localizados extraadrenalmente e têm o maior risco de metástases, enquanto que os tumores de aglomerado 2 são mais localizados adrenalmente e têm um baixo risco de metástases [4].
Diagnósticos: Metanefrinas em plasma livre são marcadores significativos
Os doentes com mutação conhecida da linha germinal, mas também com suspeita clínica de feocromocitoma/paragaganglioma (PPGL) ou se tiver sido relatado um historial de um incidentaloma adrenal ou PPGL, devem ser rastreados [4]. Numa primeira fase, devem ser recolhidos os seguintes parâmetros laboratoriais bioquímicos [1]:
- Metanefrinas em plasma livre (=metabolitos de adrenalina)
- Normetanephrines (=metabolitos de noradrenalina)
- 3-metoxitramina (=metabolitos de dopamina)
É aconselhável recolher estes dados usando a espectrometria de massa, uma vez que isto pode alcançar uma melhor sensibilidade do que outros métodos. Antes da recolha da amostra de sangue, os pacientes devem abster-se de nicotina e café, bem como de tomar SSRIs e/ou antidepressivos tricíclicos, metanfetaminas cristalinas ou cocaína, entre outras coisas. Para além dos parâmetros mencionados, é também útil determinar a cromogranina A, uma vez que é um bom marcador, especialmente para tumores assintomáticos, pelo que a ingestão de inibidores da bomba de prótons (PPI) deve ser abster-se uma semana antes do exame. A determinação do catecol por si só não é útil, disse ela, uma vez que isto poderia levar a um resultado falso-negativo. Se os valores determinados de metanefrina estiverem acima do dobro do valor normal, a imagem é indicada numa etapa de clarificação adicional [1]. Recomenda-se primeiro a RM/TC do abdómen e região pélvica; se os resultados forem negativos ou se já existir um tumor extra-adrenal/PGL, é necessária uma imagem funcional. Se o tamanho do tumor adrenal é <5 cm, O Prof. Nölting aconselha a utilização de DOPA PET/CT. DOTA-SSA-PET/CT deve ser realizado na presença de tumores extra-adrenais e de resultados negativos de imagem anatómica ou se já estiverem presentes metástases ou se o tamanho do tumor adrenal for >5 cm, explicou o orador [1].
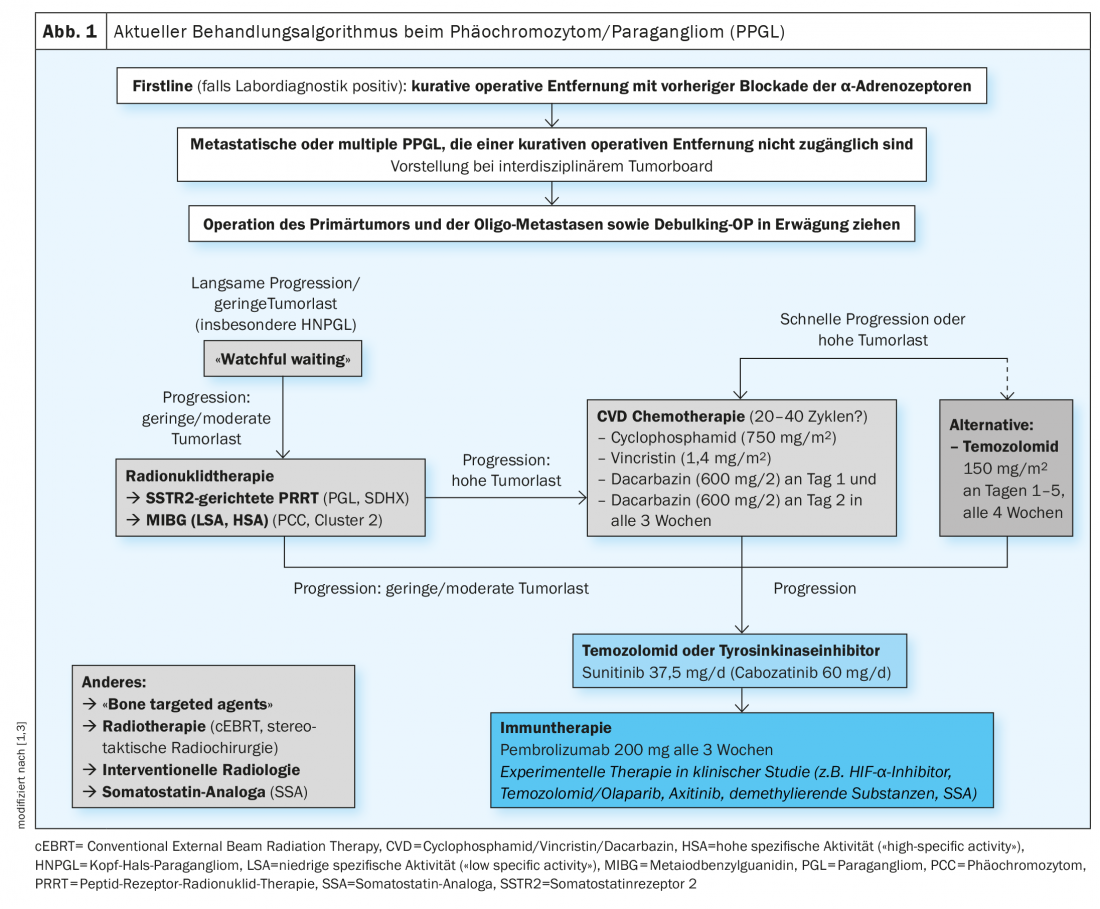
Estratégia de tratamento individualizado: novas terapias experimentais
O algoritmo de tratamento actual é mostrado na Figura 1 [1,3]. A remoção cirúrgica é considerada a terapia de primeira linha para PPGL localizada. Isto pode geralmente ser feito de forma minimamente invasiva e deve ser realizado num centro experiente [4]. Para evitar complicações, é efectuado um bloqueio medicinal dosadrenoceptores α-adrenoceptores pré-operativos. O PPGL metástático é actualmente tratado com terapia de radionuclídeos, quimioterapia ou inibidores da tirosina cinase, embora ainda não existam terapias oficialmente aprovadas. Outras novas estratégias de tratamento estão actualmente a ser testadas clinicamente. Até agora, a terapia de doenças inoperáveis ou metastáticas específicas de clusters ainda não foi estabelecida na prática clínica. Contudo, isto faria sentido para poder oferecer um tratamento personalizado e geneticamente controlado, diz o Prof. Nölting [3]. Uma publicação científica apareceu em Endocrine Reviews em 2021, na qual se discute em pormenor como pode ser desenvolvido um plano de gestão individualizado coerente para doentes com feocromocitoma/paraganglioma baseado em análises genéticas e tipagem de agregados, respectivamente, a fim de adaptar de forma óptima as opções de tratamento [3].
Congresso: Sociedade Suíça de Endocrinologia e Diabetologia 11.11.2021
Literatura:
- Nölting S: “Genetic testing in patients with pheochromocytomas/paragangliomas”, Prof. Dr. med. Svenja Nölting, SGED 11.11.2021
- Zulewski H, Grouzmann E: Diagnóstico e tratamento: “Feocromocitoma”, Swiss Med Forum 2017; 17(37): 790-796, https://medicalforum.ch/de/detail/doi/smf.2017.03057
- Nölting S, et al: Gestão personalizada do feocromocitoma e do paraganglioma. Endocr Rev 2021 Jun 19:bnab019
- Feocromocitoma – doença modelo para medicina personalizada, Dtsch Med Wochenschr 2021; 146(23): 1520-1526.
- Sociedade Alemã de Endocrinologia, Hormonas e Metabolismo: PROSPHEO, www.endokrinologie.net/sektion-nebenniere-steroide-hypertonie-7.php (acessado pela última vez em 07.12.2021)
PRÁTICA DO GP 2022; 17(1): 42-43
InFo ONCOLOGy & HEMATOLOGy 2022; 10(3): 18-19









