
Os sintomas psiquiátricos são um aspecto crucial da doença de Huntington. Uma visão geral da avaliação dos sintomas e das opções de tratamento.
Os sintomas psiquiátricos são um aspecto crucial da doença de Huntington. Também ocorrem frequentemente na fase prodromal e agravam-se com a progressão da doença [1].
A doença de Huntington, que tem uma prevalência de 10,6-13,7/100.000 habitantes no mundo ocidental, baseia-se numa desordem autossómica dominante da poliglutamina causada por um alongamento do gene Huntington no cromossoma 4 [2,3]. Quanto mais repetições, maior a severidade e menor o período de latência até à primeira manifestação dos sintomas. A doença manifesta-se clinicamente a partir de um comprimento de 39 repetições [4]. Isto resulta na degeneração de gaba e neurónios colinérgicos no neostriato e na atrofia do corpus striatum.
Os sintomas neuropsiquiátricos podem ser divididos em quatro sub-áreas: Condução, efeito, ilusão e mudanças de personalidade [5]. Pode ocorrer comportamento compulsivo, depressão, sintomas psicóticos, confabulações e desenvolvimento de demência. As deficiências na flexibilidade cognitiva e no controlo de impulsos são comuns [6].
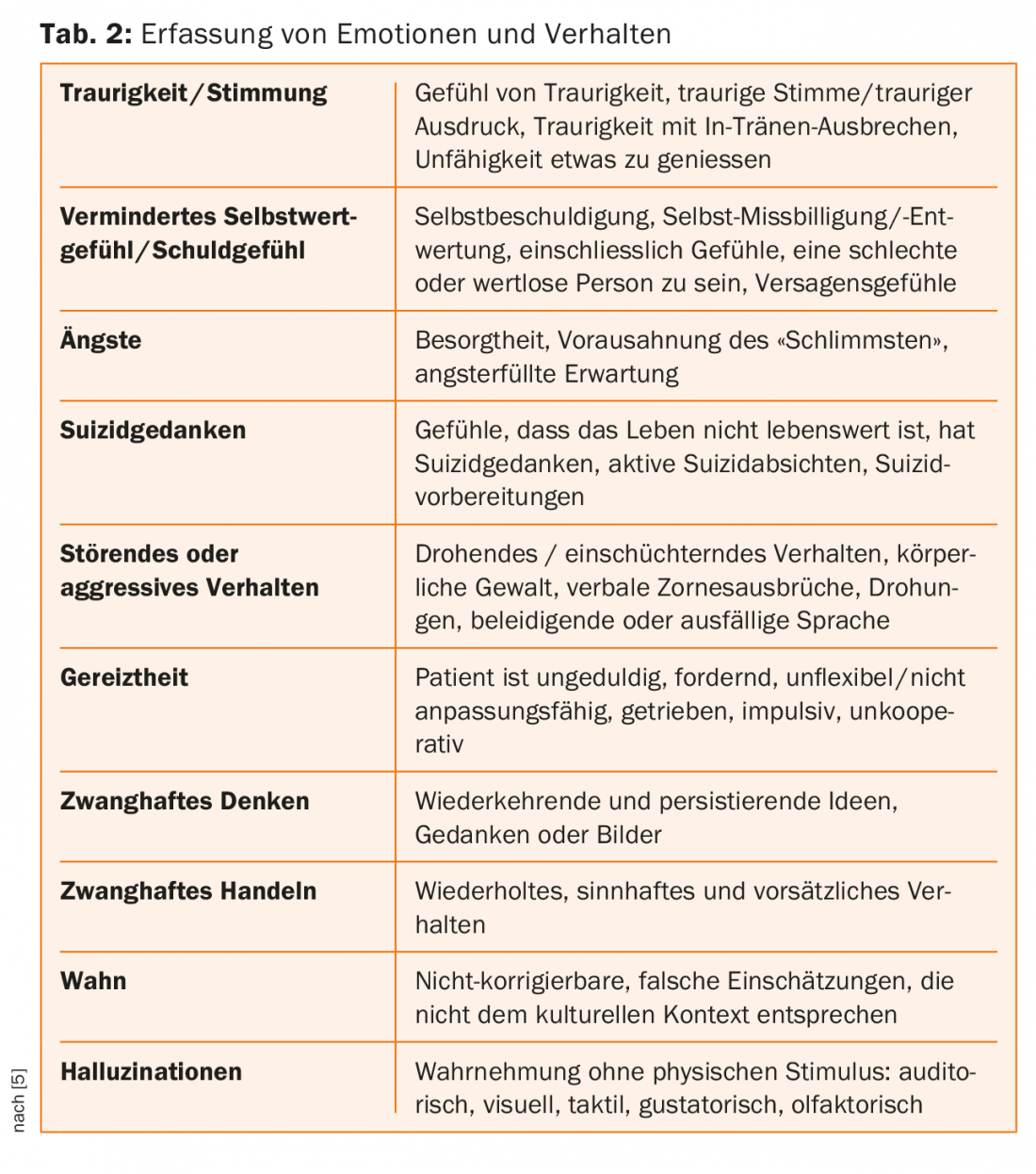
Juntamente com o registo do comportamento do doente, qualidade de vida e capacidade de funcionamento, os sintomas psiquiátricos são avaliados no padrão de exame “Unified Huntington’s Disease Rating Scale” (UHDRS) com classificações numa escala Likert de 0-4 relativamente à sua gravidade [5] – ver Quadro 1. Para além da depressão, também se pode observar um aumento da irritabilidade no decurso da doença; isto já se encontra frequentemente na fase prodromal ou na fase inicial de manifestação. A percepção mais ausente da doença é atribuída à função deteriorada no lóbulo frontal e à transmissão reduzida para o corpo de estribo [7].

Contudo, estes sintomas psiquiátricos também podem ocorrer noutras doenças neurológicas ou psiquiátricas, de modo que o diagnóstico precoce da doença de Huntington com base em sintomas psiquiátricos é difícil [1].
Os sintomas motores típicos são movimentos coréticos: involuntários, arrítmicos, contracções musculares curtas que ocorrem em regiões do corpo em mudança. Além disso, existem sintomas vegetativos tais como distúrbios do sono, perda de peso e alterações da libido [6]. Na variante clássica da doença, há precocemente – para além dos movimentos coréticos – perturbações dos movimentos oculares, distonia e impersistência motora quando se realizam acções motoras simples. As deficiências motoras reduzem a esperança de vida dos pacientes, entre outras coisas devido a quedas e pneumonia por aspiração [2].
História e situação médica
A receptora de 42 anos de idade IV foi designada para terapia de internamento pelo seu psicólogo assistente. No ambiente familiar, tinham havido conflitos crescentes devido a uma mudança na natureza do paciente que foi percebida do exterior. Isto já tinha começado há cinco anos, e a história externa da paciente mostrava-a cada vez mais irritável, verbalmente agressiva e cada vez menos capaz de lidar com a situação. Ela é propensa a explosões emocionais impulsivas e faz insinuações incompreensíveis aos membros da família. O doente tinha-se tornado “mais desconfiado” e tinha-se retirado socialmente. De manhã, ela tem cada vez mais dificuldades em levantar-se e está indiferente. Já não tinha cumprido os seus deveres domésticos e educacionais e tinha, por vezes, demonstrado um “comportamento bizarro”. O marido tinha-se mudado devido à deterioração da situação, os dois filhos comuns viviam com a cunhada. A própria paciente expressou que tinha a sensação de que a sua cunhada lhe queria tirar os seus filhos.
Como resultado de um grave acidente de bicicleta há 18 anos atrás, ela está a receber uma pensão por invalidez. Caso contrário, não eram conhecidas doenças físicas ou psiquiátricas anteriores, o historial de dependência era brando.
A paciente não tinha qualquer percepção da sua doença e quase nenhuma motivação para o tratamento. Na admissão, houve um distúrbio de mobilidade discreta e involuntária, que a paciente declarou que não se apercebeu. Quando questionado, o marido relatou um aumento dos distúrbios de movimento dos braços e das mãos, bem como do tronco. Não existem doenças psiquiátricas ou neurológicas conhecidas na família.
Descobertas clínicas
Os achados psicopatológicos mostraram uma atenção, memória e concentração reduzidas. A linha formal de pensamento foi ordenada, o pensamento estreitou-se. O paciente parecia suspeito, irritável e chocante, instável no afecto, facilmente desviável para o pólo depressivo. Havia uma ilusão paranóica em relação à cunhada, nenhuma evidência de delírios sensoriais ou perturbações do ego. Para além de uma agitação motora, o paciente era psicovegetativamente inconspícuo. Não havia indícios de perigo agudo para si ou para os outros. Neurologicamente, para além dos discretos movimentos coréticos, o estado residual após um acidente de trânsito e reconstrução plástica (plástico latissimus dorsi) da perna inferior com paresia e distúrbios sensoriais devido a uma paralisia peroneal pós-traumática foi impressionante. Houve ligeiras perturbações nas capacidades motoras finas e na coordenação, bem como um padrão de marcha instável e assimétrico com um coxear endurecedor. As sensações de pressão, temperatura e dor na perna inferior esquerda foram relatadas como tendo aumentado.
Diagnóstico e curso clínico
Um bom mês após a admissão em domo, foi feita uma apresentação na clínica neurológica do Hospital Cantonal de Lucerna com base numa suspeita de diagnóstico da doença de Huntington. Aí, o exame físico revelou apraxia do olhar, discurso estacato intermitente e discinesias da boca, mandíbula e olhos, bem como mioclonia do ângulo oral e discinesias de dedos miocloniformes bilateralmente. Além disso, houve impersistência motora da língua e das mãos (“aderência das leiteiras”), disdiadococinesia e movimentos intermitentes da coréia, bem como aumento dos reflexos musculares de todas as extremidades e um reflexo cloniforme do tendão de Aquiles bilateralmente. Ataxia de postura e marcha com um aumento da discinesia era aparente.
Os exames realizados EEG, hiperventilação e fotoestimulação, bem como a sonografia duplex dos vasos cerebrais, não mostraram quaisquer resultados patológicos. Uma ressonância magnética cMRI realizada demonstrou atrofia de caudato bilateral sem perturbações de sinal e ligeira atrofia de putaminal.
Isto reforçou a suspeita da doença de Huntington; a discinesia tardive devida a anestésicos antigos e a doença de Wilson foram consideradas como diagnósticos diferenciais.
Na clínica psiquiátrica, quetiapina 300 mg foi utilizada para tratar os sintomas afectivos e ilusórios, e o paciente foi também integrado em terapias orientadas para a acção.
Um bom dois meses após a admissão, a paciente retirou-se a seu próprio pedido e após consulta do departamento de neurologia enquanto continuava a medicação com Quetiapine retard 300 mg. No dia da descarga, foi efectuado um exame genético molecular no Hospital Cantonal de Lucerna, que forneceu provas de uma expansão CAG patológica com repetições de 46 CAG e, portanto, o diagnóstico confirmado da doença de Huntington.
Apesar dos sintomas mentais relevantes para o tratamento, ainda não era necessário tratamento anti-cinético devido aos leves sintomas motores na alta. Para além do aconselhamento genético e psicológico, foi recomendado o acompanhamento pós-internação por um serviço social e cuidados de proximidade.
Discussão
A doença de Huntington é uma doença neuropsiquiátrica que é facilmente diagnosticada nas fases tardias devido aos movimentos coréticos (ver Separador. 1-3 para registar os problemas de comportamento). No entanto, as anomalias neuropsiquiátricas não específicas ocorrem frequentemente anos antes da manifestação inicial dos sintomas motores e afectam gravemente a vida dos pacientes. Por conseguinte, é ainda mais importante incluir uma tal “doença órfã” no diagnóstico diferencial de doentes psiquiátricos anormais, para poder acompanhar e cuidar adequadamente dos doentes [8]. No nosso caso, a irritabilidade paranóica e desconfiada, a falta de controlo de impulsos, bem como o humor depressivo, para além dos movimentos coréticos que se encontravam nas fases iniciais, estavam a liderar.
Até agora, não há possibilidades de terapia causal, mas há muita investigação sobre a melhor forma de influenciar a progressão da doença [9]. As investigações laboratoriais iniciais sugerem possibilidades terapêuticas num futuro próximo [10]. Os ferries genéticos, biomarcadores ou transplantes de células estaminais estão a ser discutidos como possíveis terapias curativas, mas estas ainda não são aplicáveis, razão pela qual o diagnóstico ainda é considerado incurável [9].
Após receber o diagnóstico, o doente necessita de cuidados e aconselhamento abrangentes. Para além das terapias funcionais (ergoterapia e fisioterapia, fonoaudiologia), o início da terapia medicamentosa é indicado na presença de sintomas mentais que requerem tratamento – como no caso descrito, por exemplo, com medicamentos estabilizadores afectivos e antipsicóticos, tais como neurolépticos atípicos. Se necessário, pode ser dado tratamento com tetrabenazina ou tiapride para a hipercinesia disruptiva e L-dopa para os sintomas parkinsonoides. O aconselhamento genético, bem como o apoio social e psicoterapêutico aos pacientes e famílias afectadas, é de grande importância.

Mensagens Take-Home
- A doença de Huntington pode causar sintomas neuropsiquiátricos debilitantes durante a fase prodromal ou nas fases iniciais.
- Os sintomas neuropsiquiátricos provêm dos quatro subdomínios do impulso, do efeito, da ilusão e das mudanças de personalidade.
- O doente muitas vezes não tem qualquer percepção da doença, mesmo que os familiares e o ambiente estejam a sofrer.
- Uma avaliação dos sintomas pode ser feita, por exemplo, através do registo das anomalias comportamentais na “Unified Huntington’s Disease Rating Scale” (UHDRS) .
- Antipsicóticos atípicos e apoio psicoterapêutico podem ser utilizados no tratamento de sintomas neuropsiquiátricos.
Literatura:
- Epping EA, Kim JI, et al: Longitudinal Psychiatric Symptoms in Prodromal Huntington’s Disease: A Decade of Data (Sintomas Psiquiátricos Longitudinais na Doença de Prodromal Huntington: Uma Década de Dados). Am J Psiquiatria. 2016; 173(2): 184-192.
- Pringsheim T, Wiltshire K, et al: The incidence and prevalence of Huntington’s disease: a systematic review and meta-analysis. Desordem em movimento. 2012; 27(9): 1083-1091.
- McColgan P, Tabrizi SJ: doença de Huntington: uma revisão clínica. Eur J Neurol. 2018; 25(1): 24-34.
- Paulson HL, Albin RL: Doença de Huntington: Características Clínicas e Rotas para a Terapia. In: Neurobiologia da doença de Huntington: Aplicações à descoberta de medicamentos (eds Lo DC, Hughes RE): 2011.
- Kieburtz K: Unified Huntington’s Disease Rating Scale: Reliability and-Consistency. Distúrbios do movimento. 1996 II(2): 136-142.
- Duff K, Paulsen JS, et al: Predict HDIotHSG. Sintomas psiquiátricos na doença de Huntington antes do diagnóstico: o estudo de previsão-HD. Psiquiatria Biol. 2007; 62(12): 1341-1346.
- Hoth KF, Paulsen JS, et al: Os doentes com a doença de Huntington têm uma consciência prejudicada das suas capacidades cognitivas, emocionais e funcionais. J Clin Exp Neuropsychol. 2007; 29(4): 365-376.
- Schiefer J, Werner C, et al: Diagnóstico clínico e gestão na doença de Huntingtons precoce: uma revisão. Doença Neurológica Degenerativa e Neuromuscular. 2015; (5): 37-50.
- Lo DC, Hughes RE, editores. Neurobiologia da Doença de Huntington: Aplicações à descoberta de medicamentos. CRC Press/Taylor & Francis, 2011.
- Chen GL, Ma Q, et al.: Modulação do REST nuclear através de emendas alternativas: um alvo terapêutico potencial para a doença de Huntington. J Cell Mol Med. 2017; 21(11): 2974-2984.
Agradecimentos: Gostaríamos de expressar os nossos sinceros agradecimentos ao Prof. Dr. med. Bohlhalter e ao Dr. med. Stephan Mittas da Neurologia Lucerna, Dipl. med. Berennen-Dietrich do Rötgeninstitut Luzern e o Dr. med. Roland Spiegel da Genetica Zurich.
InFo NEUROLOGIA & PSYCHIATRY 2018; 16(1): 24-27.









