Las porfirias son un grupo de enfermedades metabólicas muy raras, determinadas genéticamente, con un trastorno de la biosíntesis del hemo. La tríada sintomática típica en una recaída consiste en dolor abdominal, síntomas cerebrales y neuropatía periférica. Otro signo importante es la decoloración roja de la orina. Para el tratamiento de un episodio agudo, la administración intravenosa de hemarginato (Normosang®) es la terapia de elección. A la hora de atender a los pacientes, es especialmente importante evitar los fármacos que desencadenan recaídas (tarjeta de emergencia). Los pacientes con porfiria recién diagnosticada deben incluirse en el Registro de Porfirias Agudas de Múnich.
Las porfirias son un grupo de enfermedades metabólicas muy raras, determinadas genéticamente, que se basan en un trastorno de la biosíntesis del hemo. En el organismo humano, el hemo es un componente de la hemoglobina, responsable del transporte de oxígeno, y de la mioglobina, proteína de almacenamiento de oxígeno, en la que aparece como grupo prostético portador de hierro, así como de numerosos citocromos, que intervienen en el metabolismo energético de prácticamente todas las células de la cadena respiratoria . Para el recambio continuo de estas proteínas, el hemo debe ser suministrado constantemente.
En la biosíntesis del hemo intervienen ocho enzimas; la regulación de la síntesis se produce mediante la inhibición por retroalimentación de la δ-ALA sintasa, la primera enzima de la biosíntesis, por el producto final hemo. Existen dos isoenzimas en el organismo: la δ-ALA sintasa 1 se da en todos los tejidos, la δ-ALA sintasa 2 sólo en la médula ósea. Debido a su importancia cuantitativa, el hígado y la médula ósea son los lugares más importantes para la síntesis del hemo.
Fisiopatología de las porfirias
Las mutaciones hereditarias (o raramente espontáneas) en los genes de las enzimas de la biosíntesis del hemo provocan una pérdida parcial de la función de la enzima codificada. Esta pérdida parcial de función (es decir, hasta el 50%) se compensa suficientemente con la estimulación de las δ-ALA sintasas, por lo que normalmente no hay deficiencia de hemo. Por lo tanto, los portadores de la predisposición genética no suelen tener problemas con el rendimiento metabólico reducido de la enzima afectada. La situación se vuelve problemática cuando aumentan las necesidades de hemo, por ejemplo al tomar un fármaco como la fenitoína, que induce en el hígado enzimas de degradación del citocromo P450 que contienen hemo. Entonces se estimula muy fuertemente la síntesis de hemo y se produce un “atasco” en el punto estrecho del defecto enzimático debido a la reducción de la actividad al 50%: Los intermediarios del metabolismo del hemo (especialmente los precursores de la porfirina porfobilinógeno y δ-aminolevulinato) se acumulan en las células, entran en el espacio extracelular y se distribuyen por el organismo.
Tres formas de porfiria
Según cuál sea la enzima afectada en las porfirias agudas, se distingue entre la variante frecuente “porfiria aguda intermitente” (AIP) y las variantes más raras “porfiria variegata” (PV) y “coproporfirinuria hereditaria” (HKP) (Fig. 1).
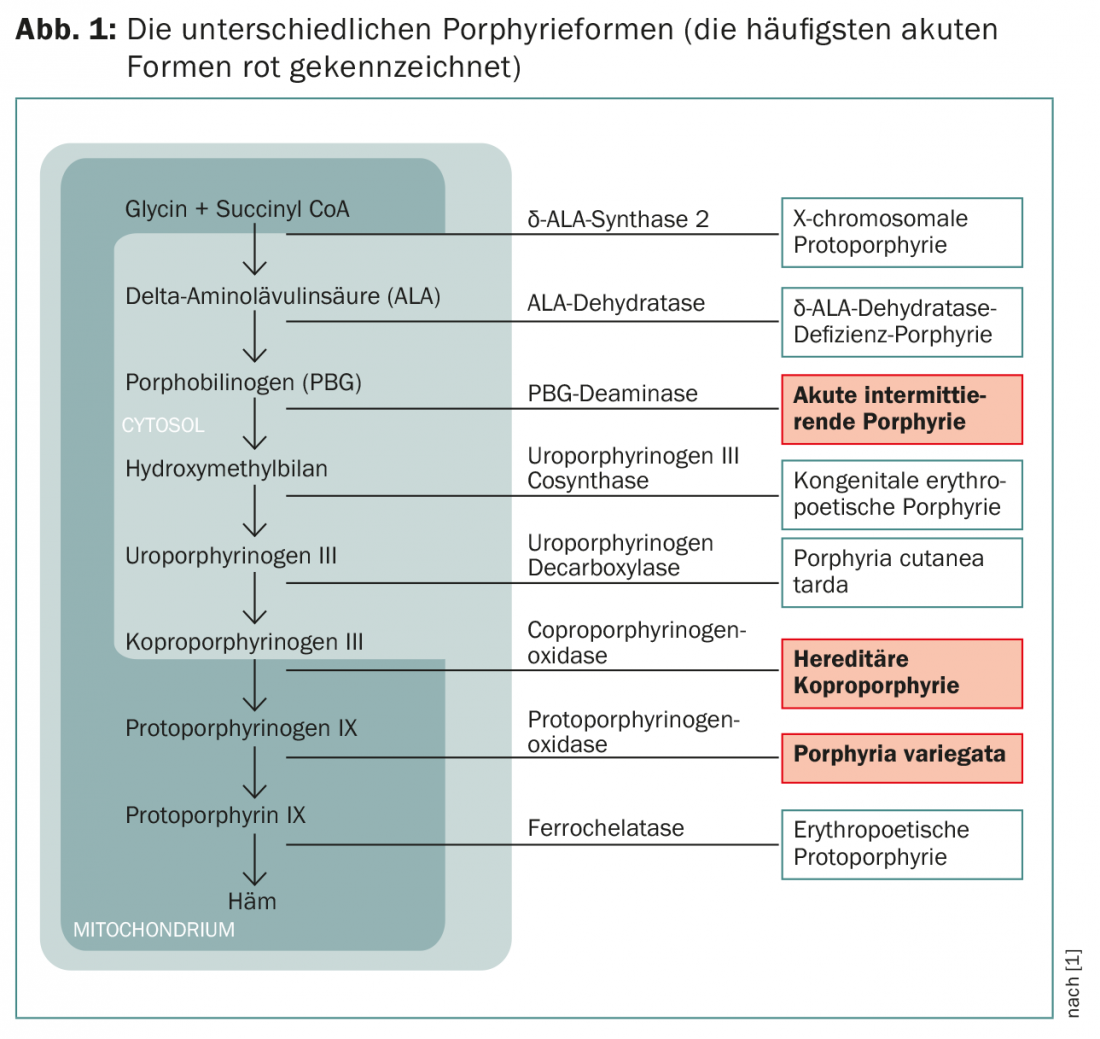
Los síntomas dependen de los metabolitos que se acumulen. Los precursores de la porfirina, que siguen siendo hidrosolubles, penetran en los nervios y provocan síntomas neurológicos; las porfirinas liposolubles se depositan en la piel y provocan síntomas cutáneos por la exposición a la radiación UV. Estas dolencias son muy variadas y a menudo confusas, ya que también se presentan con un gran número de otras enfermedades. Por lo tanto, el diagnóstico correcto suele realizarse en una fase correspondientemente tardía.
Síntomas: Dolor abdominal, orina roja, trastornos neurológicos
En las tres formas, la aparición aguda de síntomas se denomina recaída. Las recaídas pueden ser leves y entonces están determinadas principalmente por el dolor abdominal. Se producen repetidamente sin que se logre hacer el diagnóstico correcto. Sólo en el caso de un episodio grave en el que, además del dolor abdominal, aparezcan signos de síntomas centrales (como estado de ánimo depresivo, alucinaciones, coma) y síntomas neurológicos periféricos (dolor de espalda, parálisis) (tríada sintomática), aumenta la probabilidad de que se realice el diagnóstico correcto (Fig. 2).
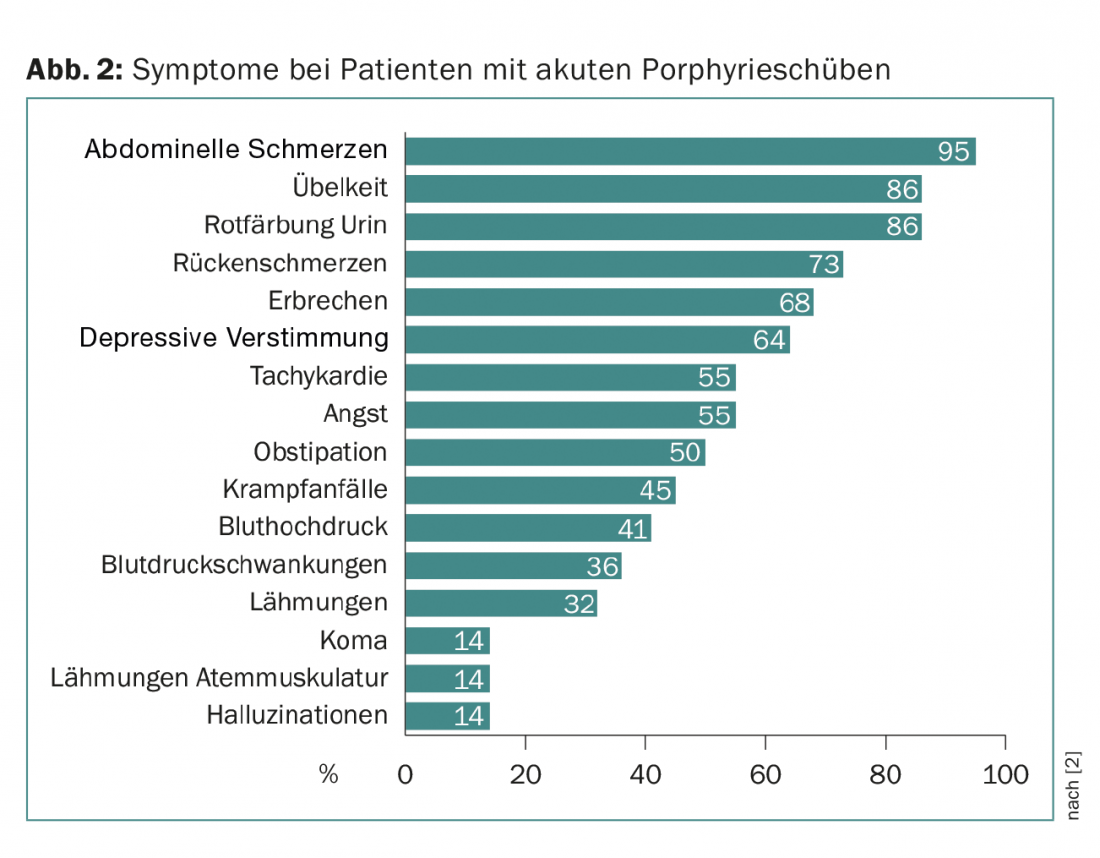
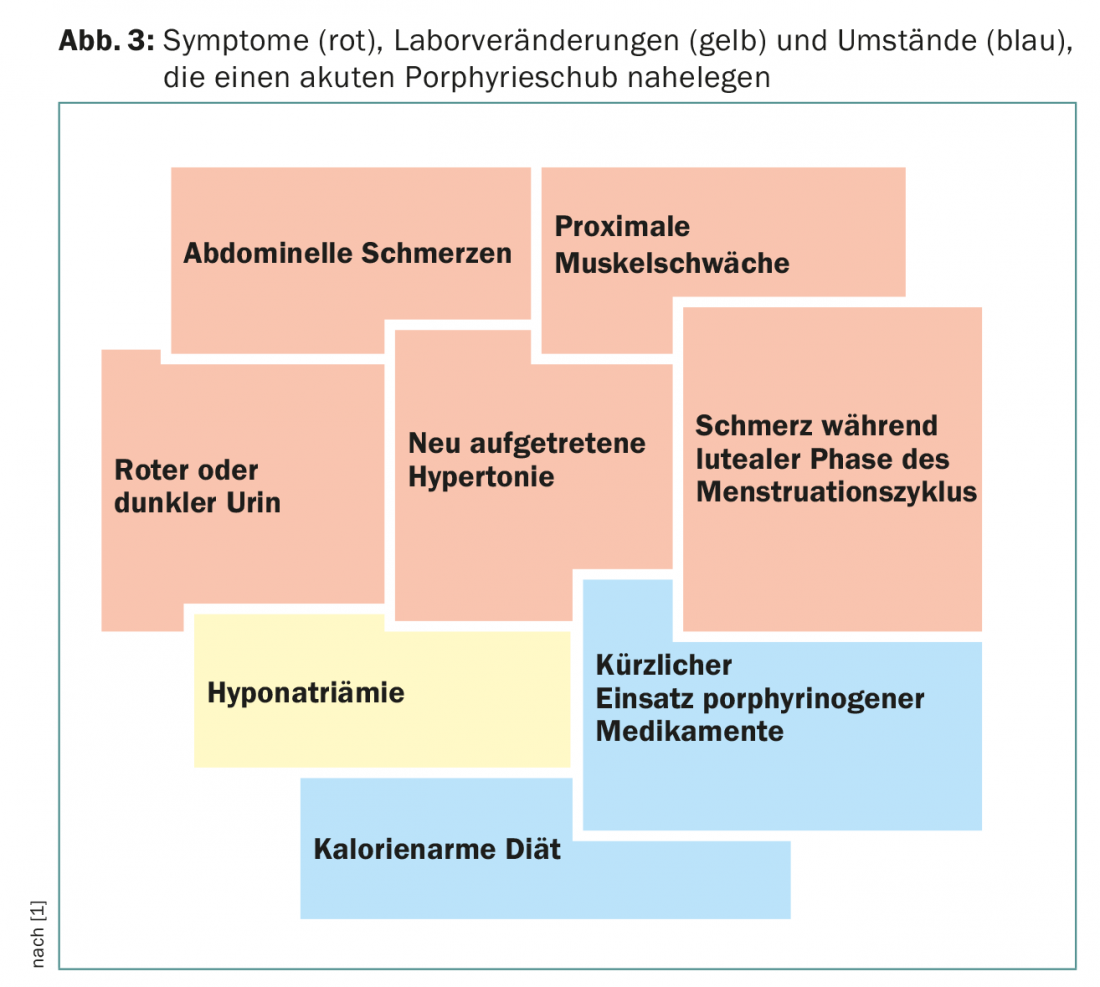
Un importante signo clínico adicional es la decoloración roja de la orina, causada por la autooxidación del porfobilinógeno a porfobilina. Junto con cambios de laboratorio como la hiponatremia, estos síntomas sugieren una porfiria aguda (Fig. 3). Los pacientes suelen ser atendidos en unidades de cuidados intensivos debido a la gravedad de los síntomas, y a menudo es un asistente joven el que recuerda la enfermedad rara por su proximidad al examen de estado.
La hiponatremia puede provocar convulsiones, que no pocas veces se tratan con carbamazepina o fenitoína, que figuran entre los fármacos que favorecen las convulsiones, sin conocer el diagnóstico. Los síntomas cerebrales pueden ir desde un deterioro mental leve hasta la psicosis paranoide, el delirio o el coma. En casos aislados, se produce una mono o hemiparesia central y, en caso de parálisis respiratoria, la necesidad de ventilación mecánica es inminente, por lo que puede desarrollarse una situación de riesgo vital. En el síndrome de encefalopatía posterior reversible (PRES), detectado mediante una resonancia magnética, puede producirse ceguera cortical con pérdida completa de la agudeza visual.
Algunos pacientes desarrollan neuropatía periférica, predominantemente motora, durante la recaída. En una gran proporción de los afectados, la polineuropatía se desencadena por medidas iatrogénicas (administración de fármacos porfirinógenos) ignorando la presencia de porfiria aguda. La neuropatía tiene una distribución atípica, es decir, los brazos están más gravemente afectados que las extremidades inferiores, los músculos proximales más pronunciados que los grupos musculares distales. En un tercio de los pacientes, la parálisis comienza en las piernas, en la mitad con énfasis proximal. La distribución asimétrica de la paresia es frecuente. Los signos de neuropatía sensorial son alteraciones sensoriales en forma de calcetín y guante o en la zona del tronco como un “traje de baño”. En las porfirias agudas más raras, la PV y la HKP, también se producen síntomas cutáneos. Esto se debe a que no sólo se elevan los precursores de la porfirina, sino que, debido a la posición del defecto enzimático responsable, también se depositan en la piel productos intermedios posteriores de la síntesis de porfirina.
Diagnóstico
El aumento de la detección de precursores de porfirinas (δ-ALA y PBG) en la orina y no -como aún se supone erróneamente- principalmente de porfirinas sugiere la sospecha de porfiria aguda. El porfobilinógeno se mide primero de forma semicuantitativa en una prueba rápida. El problema es que esta prueba no está disponible en muchos laboratorios. Si la prueba es positiva, se procede al análisis cuantitativo de PBG y δ-ALA: si el resultado está relacionado con el valor de creatinina en orina, suele bastar con una muestra de orina espontánea. La orina de 24 horas ya no es necesaria.
Si los precursores de porfirina están elevados, la presencia de porfiria aguda intermitente se confirma mediante la determinación de la PBG deaminasa en los eritrocitos y, si está disminuida, mediante un análisis de mutación (prueba genética). Para el diagnóstico de las otras dos porfirias agudas, se requiere un análisis de orina y heces para detectar uroporfirinas y coproporfirinas, respectivamente.
Hoy en día, la confirmación final del diagnóstico se realiza a través de más de 300 mutaciones diferentes en el gen de la PBG deaminasa, es decir, prácticamente una de cada dos posiciones de aminoácidos puede estar afectada por una mutación. Por lo tanto, siempre debe secuenciarse el gen completo en el diagnóstico inicial. Si se conoce la mutación, es posible realizar pruebas específicas a los familiares para detectar la presencia de esta mutación.
Sólo alrededor del 10-15% de los portadores del gen desarrollan una manifestación de porfiria a lo largo de su vida. Esto se atribuye, por una parte, a que la confrontación con los agentes desencadenantes no se produce en conocimiento de la predisposición y, por otra, a que los genes modificadores protegen contra la manifestación de una recaída.
Opciones terapéuticas
Anteriormente, la única terapia disponible era la infusión de altas dosis de glucosa; esto inhibe la síntesis de hem mediante la regulación a la baja de la actividad de la δ-ALA sintasa. Hoy en día, la administración intravenosa de hemarginato (Normosang®) es la terapia de elección para “frenar” la biosíntesis del hemo. El fármaco se administra como una infusión corta durante cuatro días; una mezcla con albúmina humana es útil debido al efecto irritante de la vena. Los pacientes individuales que sufren episodios crónicos recidivantes reciben infusiones a través de un port-a-cath. Está claro que los fármacos que favorecen las recaídas deben interrumpirse y no deben volver a utilizarse. Los fármacos clásicos que desencadenan las recaídas son, por ejemplo, los anticonvulsivos fenitoína, carbamazepina, oxcarbazepina y ácido valproico, los barbitúricos, los analgésicos metamizol y fenilbutazona, los antibióticos sulfonamidas y el antifúngico griseovulvina. El lorazepam, el diazepam, la morfina, la petidina, la clorpromazina y el ondansetrón, por ejemplo, se consideran seguros.
Los pacientes reciben una tarjeta de emergencia, que también remite a la página web www.drugs-porphyria.com, donde se actualizan constantemente las listas de medicamentos recetados y seguros. En casos individuales, es difícil llevar a cabo la asignación con certeza.
Atención interdisciplinar
Hoy en día, el diagnóstico y la terapia de los pacientes con porfirias agudas se llevan a cabo en un equipo interdisciplinar con hematólogos, gastroenterólogos, neurólogos, anestesistas, médicos de laboratorio, dermatólogos, ginecólogos y genetistas [1]. Los siguientes aspectos son de especial importancia:
- Recaídas crónicas recurrentes
- Tratamiento de complicaciones neurológicas graves relacionadas con la recaída
- Preparación y realización de la anestesia
- Evitar diagnósticos erróneos
- Cuidados de las pacientes embarazadas con porfiria
- Diagnóstico diferencial de posibles alteraciones cutáneas
- Fiabilidad del análisis de mutaciones.
Nuevo diagnóstico: inclusión en el registro de porfiria
Debido a la rareza de las enfermedades, la información sobre el curso, la gravedad de las manifestaciones clínicas y el deterioro de la calidad de vida, los posibles efectos secundarios de los tratamientos, etc. sólo puede obtenerse de los registros. En el registro de porfirias agudas de Múnich se han documentado 60 pacientes con porfirias agudas hasta febrero de 2016. Los pacientes recién diagnosticados deben ser notificados a este registro [2]. Se está construyendo una página web al respecto (www.akuteporphyrie.de).
Literatura:
- Petrides PE (ed.), et al: Las porfirias agudas, folleto informativo para médicos. 4ª edición 2015 Munich (disponible en Orphan Europe Ulm).
- Bronisch O, et al.: Porfiria aguda intermitente en Alemania: análisis provisional de 45 pacientes de una única institución (Múnich). Congreso Internacional sobre Porfirinas y Porfirias, Düsseldorf, 9/2015.
InFo ONcOLOGíA & HEMATOLOGíA 2016; 4(2): 16-19