
Les traitements symptomatiques et l’évitement des facteurs de provocation restent au cœur de la thérapie. Les options thérapeutiques moléculaires, potentiellement curatives, de l’épidermolyse bulleuse sont encore au stade de la recherche. L’évaluation diagnostique de cette maladie auto-immune géno- et phénotypiquement hétérogène doit tenir compte d’un certain nombre d’éléments.
L’épidermolyse bulleuse (EB) est un groupe de génodermatoses hétérogènes d’un point de vue génotypique et phénotypique, caractérisées par une fragilité mécanique excessive des tissus épithélialisés. Avec une prévalence d’environ 500 000 cas dans le monde, l’EB est une maladie rare [1,2]. A ce jour, des mutations ont été décrites dans plus de 20 gènes codant pour des composants impliqués dans la construction des filaments de kératine du cytosquelette, des molécules d’adhésion, des desmosomes, des hémidesmosomes et des fibrilles d’ancrage des épithéliums. (Fig. 1). Par conséquent, l’intégrité structurelle et fonctionnelle de l’adhésion intraépidermique et de l’adhérence dermoépidermique à la peau et aux muqueuses est perturbée, ce qui affecte la fonction de barrière, l’interaction cellule-cellule et cellule-matrice, la prolifération, la régénération tissulaire et les processus de différenciation. [3–5]. Le spectre combinatoire du type, de l’homozygotie ou de l’hétérozygotie, du nombre (transmission mono ou digénique) et de la localisation de la mutation dans le segment de gène concerné, ainsi que la perturbation quantitative (absence ou réduction) et qualitative (perte graduelle de fonction) de l’expression des protéines qui en résulte, détermine des variances géno- et donc phénotypiques considérables de l’EB. Outre le défaut génétique primaire, des facteurs épigénétiques et biochimiques secondaires (par exemple, l’induction de cascades inflammatoires chroniques et le remodelage tissulaire), ainsi que des facteurs environnementaux, influencent la gravité clinique [6,7].
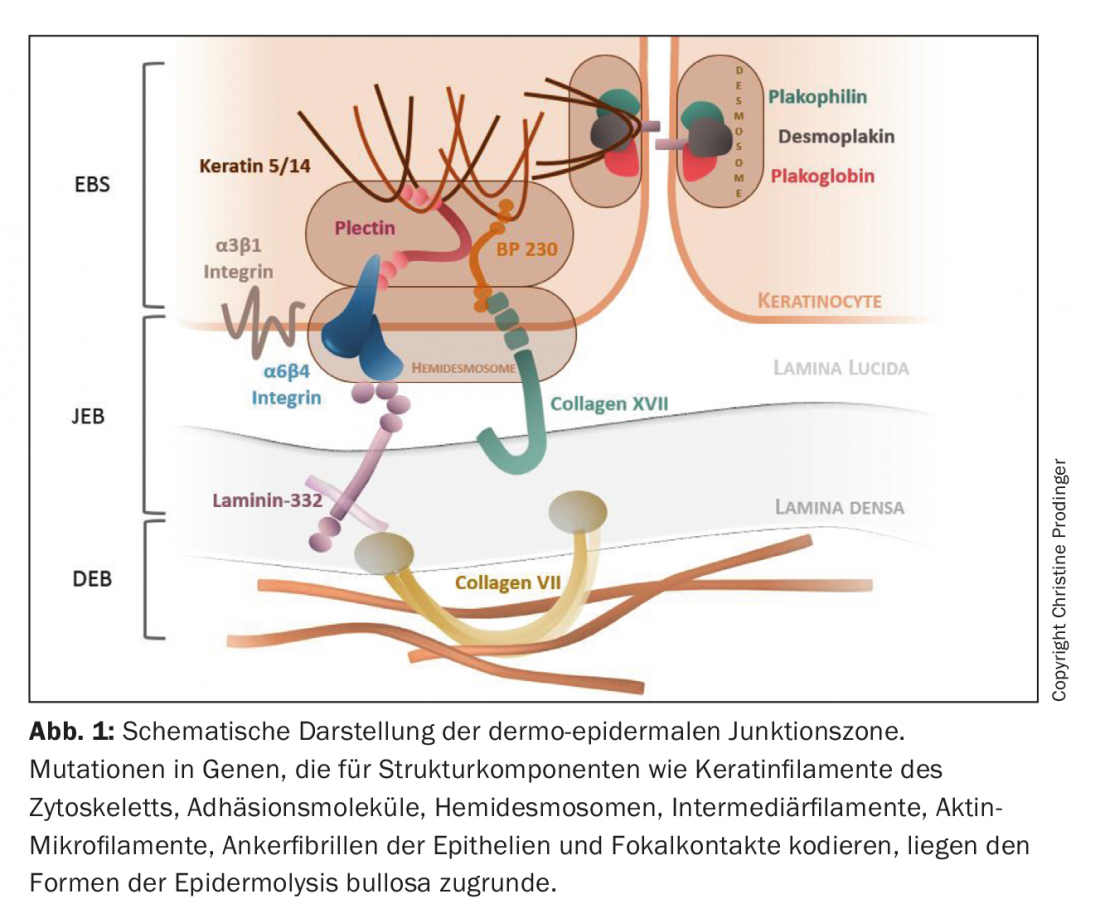
Comme les gènes index concernés ne sont pas seulement exprimés dans la peau et les muqueuses proches de la peau, mais également dans d’autres épithéliums (tractus respiratoire, urogénital et gastro-intestinal) et dans les tissus mésenchymateux (muscles squelettiques), des manifestations extracutanées primaires sont également possibles dans ces régions. L’EB peut ainsi se transformer en une maladie systémique avec une morbidité et une mortalité significatives [8].
Classification physiopathologique
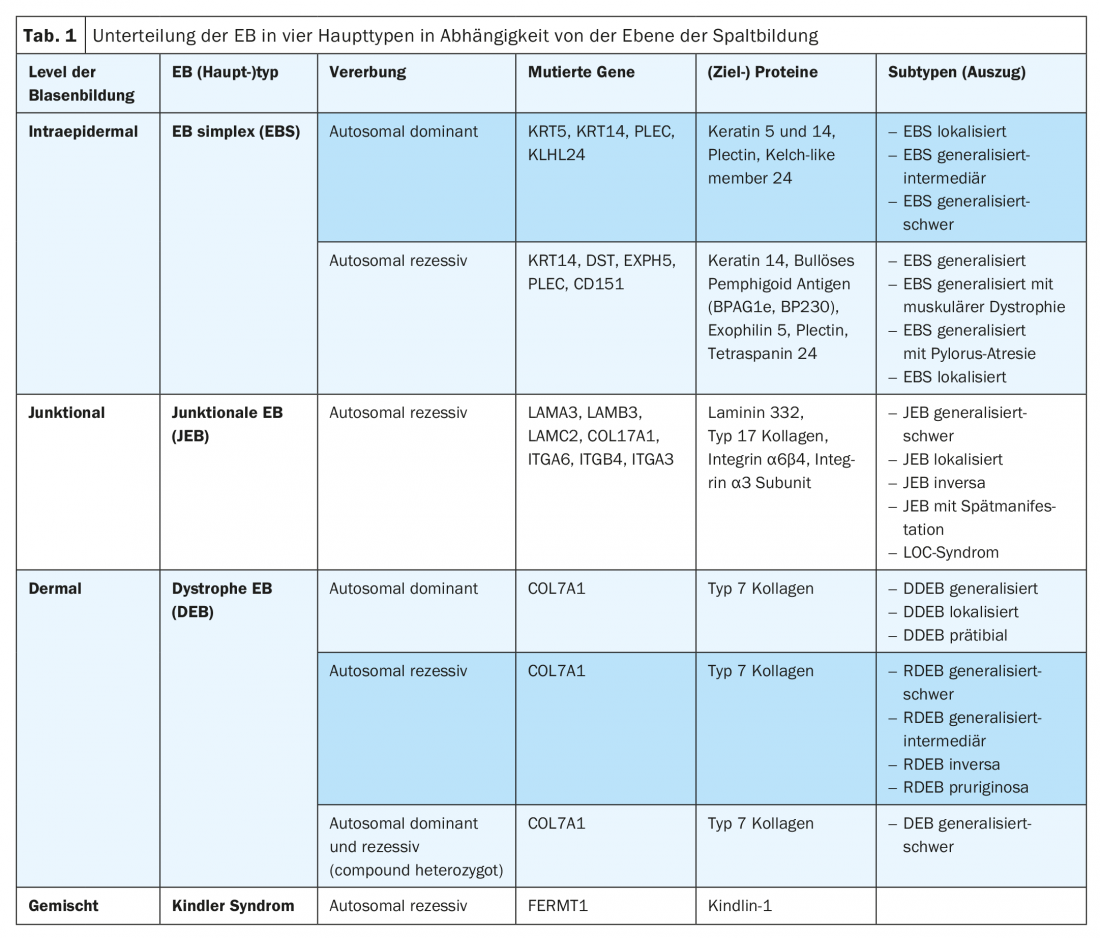
Le principal symptôme de l’EB est la fragilité accrue à excessive de la peau et des muqueuses face aux contraintes mécaniques, qui varie en fonction du sous-type, avec formation de cloques, d’érosions, d’ulcérations, de croûtes ou de cicatrices. En fonction du niveau de formation de la fente en tant que fonction et conséquence de la mutation sous-jacente, l’EB est divisée en quatre types principaux (tableau 1) [9]. En raison de la disponibilité croissante de techniques modernes de diagnostic moléculaire (par exemple, le “séquençage de nouvelle génération”), de nouveaux (sous-)types d’EB sont également régulièrement décrits. Ainsi, une mutation dans le gène KLHL24 codant pour un composant du complexe ubiquitine-ligase a pu être attribuée à une variante autosomique dominante de l’EB simplex, qui entraîne une ubiquitination et une dégradation excessives de la kératine 14 [10–12]. De plus, dans un phénotype similaire au syndrome de Kindler, une mutation a récemment été identifiée dans le gène CD151, qui code pour une tétraspanine dans la zone de la membrane basale. Cette protéine transmembranaire interagit avec les intégrines et est impliquée dans les processus de croissance, de développement et de motilité cellulaires [13]. De plus, une mutation a été récemment découverte dans le gène PLOD3, qui code pour la lysyl hydroxylase 3 et régule le processus post-traductionnel du collagène de type 7. Cliniquement, les personnes atteintes présentent des défauts étendus du tissu conjonctif, des contractures articulaires, des malformations squelettiques et un retard de croissance. Le niveau de formation des bulles est similaire à celui de l’EB dystrophique récessive dans la sublamina densa [14].
Algorithme de diagnostic
En particulier chez le nouveau-né présentant des bulles, il faut d’abord exclure une origine traumatique, métabolique, hématologique, infectieuse, médicamenteuse ou auto-immune [15]. Par la suite, un algorithme de diagnostic est appliqué :
- Anamnèse (familiale) et clinique
- Détermination de la contamination microbienne (par ex. prélèvements, PCR, sérologie)
- Biopsie périlésionnelle pour l’évaluation histologique d’éventuels diagnostics différentiels
- Immunofluorescence directe paralésionnelle pour déterminer le niveau de clivage et l’expression protéique semi-quantitative d’une bulle induite (par exemple, rotation d’une gomme de crayon jusqu’à l’apparition d’un érythème) et fraîche (pas plus de 12 heures) sur une peau non exposée au soleil.
- Microscopie électronique à transmission pour déterminer le plan de clivage et les défauts morphologiques
- Analyse des mutations (selon le cas, comme séquençage du génome entier, de l’exome, des clusters, des panels)
Le (sous-)type d’EB peut être déterminé en fonction des résultats obtenus. Le plan de clivage déterminé par microscopie électronique à transmission et immunofluorescence définit le type principal d’EB. Le phénotype clinique est défini en indiquant la sévérité relative (légère, intermédiaire, sévère) et le schéma de distribution (localisé, généralisé) – le cas échéant, les symptômes caractéristiques tels que la pseudosyndactylie sont également mentionnés. Outre les résultats spécifiques obtenus par microscopie électronique à transmission ou par cartographie par immunofluorescence, la protéine spécifique et le gène concerné, le type de mutation ou la mutation spécifique sont indiqués [9]. Les succès obtenus dans la caractérisation moléculaire des bases pathogéniques des différents types d’EB permettent en outre un conseil génétique, un pronostic et un diagnostic prénatal plus précis et constituent une condition préalable à des interventions thérapeutiques ciblées innovantes.
Principes thérapeutiques
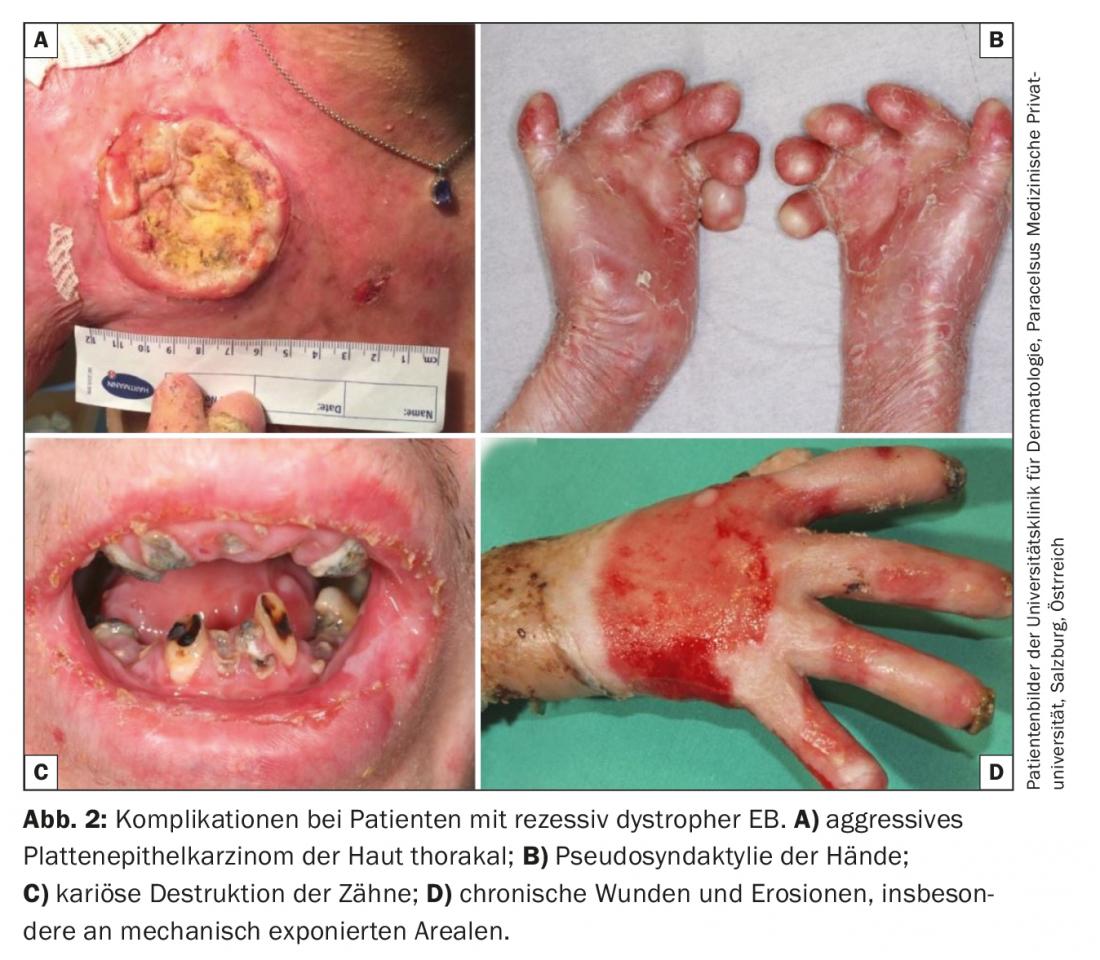
En l’absence d’options thérapeutiques curatives dans la pratique clinique quotidienne, le traitement de toutes les formes d’EB se concentre sur l’évitement des facteurs de provocation et l’optimisation des approches thérapeutiques symptomatiques. Ces mesures comprennent un traitement local adéquat et adapté au stade de la plaie, l’amélioration ou le maintien de la barrière cutanée par des soins cutanés optimisés, le soulagement de la douleur et des démangeaisons, la prévention et le traitement des infections (traitement antiseptique et antibiotique intermittent), le maintien d’un apport calorique et nutritionnel suffisant et, enfin, la prévention et le traitement des complications telles que l’anémie, l’ostéoporose et les carcinomes épidermoïdes à évolution agressive. (Fig. 2) [16,17].
Les perspectives des options thérapeutiques moléculaires, potentiellement curatives, dans l’EB sont en principe encourageantes, même s’il est encore difficile d’évaluer leur mise en œuvre large, sûre, (durablement) efficace et pratique dans la pratique clinique quotidienne pour les patients EB. L’éventail des méthodes est varié :
Thérapie génique : en 2006, la première thérapie génique par transplantation de kératinocytes corrigés du gène LAMB3 a été réalisée chez un patient atteint de JEB autosomique récessive (mutation du gène LAMB3) [18]. Dans cette thérapie de “remplacement de gène”, des cellules de peau cultivées ont été transfectées avec un vecteur contenant une copie fonctionnelle d’ADNc du gène muté causal et transplantées en tant qu’équivalents épithéliaux de peau sur des zones de plaies. La zone de jonction dermo-épidermique est restée structurellement et fonctionnellement stable pendant les 14 années de suivi et ne présente aucun signe de cloquage, d’inflammation ou de réaction immunitaire contre le néoantigène introduit dans le cadre du traitement. Dans une étude récente, des kératinocytes autologues ont été cultivés selon le même principe, des cellules souches épidermiques ont été isolées et ont subi une correction génétique ciblée, puis ont été expansées et transplantées sur de larges zones de plaies chez un garçon de 7 ans atteint de JEB (mutation du gène de la laminine 332). Cela a permis d’obtenir une réépithélialisation sur 80% de la surface du corps. La réponse clinique a été corrélée à l’expression de la laminine 332, qui se poursuit à ce jour [19]. L’efficacité est limitée par un nombre (trop) faible de cellules souches épidermiques dans les cultures primaires des patients EB pour assurer une correction permanente, qui sont notamment déplétées par des plaies chroniques et avec l’âge, ainsi que par des aspects de sécurité (concernant les vecteurs viraux et la génotoxicité ou la mutagénèse par insertion induite). Des développements technologiques sont en cours d’évaluation dans le cadre d’études en cours et devraient améliorer le profil risque/bénéfice de cette méthode, qui est particulièrement adaptée aux plaies chroniques circonscrites, fortement symptomatiques ou à risque de complications à long terme [20].
Le “gen silencing” pour les formes autosomiques dominantes d’EB (c’est-à-dire l’inactivation de l’allèle muté avec des “small interfering RNAs”), ainsi que des approches topiques de thérapie génique faciles à appliquer, sont également en cours de développement. Dans le cas de ces derniers, par exemple Les vésicules extracellulaires, isolées à partir de cellules souches mésenchymateuses allogéniques, sont capables de transporter la protéine de collagène de type 7 manquante et l’ARNm COL7A1 vers les cellules cibles [21, 22].
Les technologies d’édition du génome qui utilisent les propriétés des nucléases programmables (par ex. CRISPR/Cas9, TALEN, nucléases à doigt de zinc) sont basées sur la modification/correction de séquences de gènes mutés. Les séquences mutées sont supprimées et des séquences étrangères correctives sont insérées ou des bases individuelles sont corrigées par des mécanismes de réplication cellulaires. Cependant, les préoccupations en matière de sécurité, notamment en ce qui concerne la précision insuffisante et les sites de liaison potentiels hors cible, ne justifient pas actuellement une application (in vivo) chez l’homme [23,24].
Un phénomène clinique remarquable qui peut se produire dans toutes les formes principales d’EB est l’apparition (spontanée) de zones de peau saine sans cloques suite à des événements génétiques correctifs, ce que l’on appelle le mosaïcisme réversible ou “thérapie génique naturelle”. Les tentatives de transplantation de kératinocytes réversibles obtenus par biopsie à l’emporte-pièce dans le cadre d’essais cliniques de phase I sur des plaies EB affectées après expansion in vitro n’ont jusqu’à présent pas répondu aux attentes, principalement en raison de la perte progressive des cellules réversibles [25,26].
Thérapie cellulaire : les premiers résultats d’une transplantation de cellules souches de moelle osseuse ont montré, malgré l’absence de preuve d’une concentration de collagène de type 7 restitué, la présence de cellules donneuses dans la peau (cellules souches de moelle osseuse pluripotentes présumées reprogrammées) et une bonne réponse, parfois pendant plusieurs années, chez certains patients atteints d’EB récessive dystrophique. Ce succès, dont le mécanisme sous-jacent n’est pas encore clair, a été atténué par une augmentation significative de la mortalité périprocédurale. Des thérapies de conditionnement et des protocoles de transplantation révisés devraient améliorer la tolérance [27,28].
Les substrats des thérapies cellulaires alternatives comprennent les cellules stromales mésenchymateuses allogéniques appliquées par voie intradermique ou intraveineuse et les cellules souches mésenchymateuses adipogènes. Une meilleure cicatrisation des plaies et une réduction des signes d’inflammation de la peau ont ainsi pu être observées, du moins temporairement, ce qui est toutefois probablement dû en premier lieu à l’induction de processus immunomodulateurs favorables [29]. Un effet clinique a également déjà été démontré dans les premières études avec des fibroblastes autologues de type sauvage ou génétiquement corrigés injectés par voie intradermique, qui produisent également du collagène de type 7 en plus des kératinocytes [30,31].
Des cellules similaires aux cellules souches embryonnaires pluripotentes peuvent également être obtenues à partir de cellules somatiques (par exemple, des fibroblastes ou des kératinocytes) par transfection de trois ou quatre facteurs de transcription embryonnaires. L’utilisation de ces cellules souches pluripotentes induites (iPSC), qui peuvent à nouveau se différencier en différents types de cellules (par ex. kératinocytes ou fibroblastes), est également étudiée dans l’EB dans le cadre d’études précliniques. L’utilisation de cellules/kératinocytes révertants pour la production d’iPSC présente un grand potentiel, car elle permet de renoncer à la correction génétique et aux risques qui en découlent [32,33].
Les thérapies protéiques : On tente également de corriger la zone de jonction dermo-épidermique défectueuse en substituant la protéine manquante ou mal produite. Alors que les études sur l’utilisation du collagène recombinant de type 7 en sont encore au stade préclinique, l’application topique d’un gel contenant un virus de l’herpès simplex de type 1 exprimant le collagène de type 7 modifié, par exemple, fait déjà l’objet d’essais cliniques [34,35].
Thérapies basées sur l’ARN/”Petites molécules” : Les mutations non sens, qui provoquent un codon d’arrêt par une mutation ponctuelle de l’ADN et qui interrompent ainsi la traduction, sont responsables d’environ 10% de toutes les maladies génétiques humaines. En présence de cette mutation, des médicaments tels que les aminoglycosides (par exemple la gentamycine) ou l’immunomodulateur amlexanox peuvent entraîner une “surlecture” (read-through) du codon stop (par exemple la mutation COL7A1) en se liant aux ribosomes, permettant ainsi la production de protéines fonctionnelles [36].
Des modifications au niveau de l’ARN sont également obtenues par “saut d’exon médié par oligonucléotide antisens” (suppression ciblée d’exons contenant des mutations) et par “trans-splicature d’ARN médiée par épiceosome” (SMaRT) (correction de segments pré-ARN mutés). Cette dernière technologie a déjà permis de corriger avec succès en préclinique une mutation du gène de la pectine dans EB simplex et une mutation COL7A1 dans EB dystrophique récessive, ainsi que des mutations autosomiques dominantes dans le gène de la kératine 14 d’une lignée de cellules EB simplex [37,38].
Des “petites molécules” sont utilisées comme médiateurs d’un effet “disease-modifying”. Il s’agit notamment du calcipotriol topique, qui est censé renforcer les défenses antimicrobiennes endogènes et améliorer la cicatrisation des plaies en augmentant l’expression du peptide antimicrobien cathelicidine. [39] ainsi que la diacéréine topique, un composant de la racine de rhubarbe et un inhibiteur puissant de l’IL-1β pro-inflammatoire, qui, selon des données publiées provisoirement, est capable de réduire la formation de bulles chez les patients atteints de SEP [40]. Le rigosertib, un inhibiteur oral de la tyrosine kinase, présente également une inhibition sélective in vitro des cellules tumorales épithéliales de la plaque chez les patients atteints d’EB dystrophique récessive, et son efficacité et sa sécurité sont en cours d’évaluation dans une étude en cours. Carcinomes épidermoïdes très agressifs en tant que complication de l’EB chronique Les plaies constituent une cause majeure de décès, en particulier chez les patients atteints d’EB dystrophique récessive [41]. Outre le rigosertib, le nivolumab, un anticorps monoclonal antirécepteur PD-1, fait actuellement l’objet d’une évaluation contrôlée de son efficacité dans les carcinomes épidermoïdes localement avancés et métastatiques dans la cohorte EB (EudraCT 2016-002811-16).
Messages Take-Home
- L’épidermolyse bulleuse (EB) est une maladie géno- et phénotypiquement hétérogène. Les formes graves évoluent vers une maladie multisystémique avec une morbidité et une mortalité prononcées. Les principales causes de décès sont les infections, la dystrophie, la défaillance des organes et les carcinomes épidermoïdes. Ces dernières apparaissent tôt et de manière multiple dans les plaies chroniques et présentent une évolution agressive.
- Le diagnostic est établi en corrélation avec la clinique, l’histologie, l’immunofluorescence et l’analyse moléculaire. Malgré les approches innovantes, parfois causales, des stratégies thérapeutiques moléculaires, la guérison est une perspective d’avenir indirectement encore vague. L’immunomodulation est une stratégie thérapeutique prometteuse.
- Les composants épigénétiques et biochimiques secondaires ainsi que les facteurs environnementaux, qui peuvent par exemple induire des cascades inflammatoires chroniques et systémiques, ont une grande pertinence pathogénique. Comme pour d’autres maladies rares, certaines caractéristiques rendent difficile la réalisation d’essais cliniques et donc la production de données probantes de grande qualité.
Littérature :
- Fine JD : Epidermolyse bulleuse inhérente. Orphanet J Rare Dis 2010 ; 5 : 12.
- Fine JD, et al : Epidermolysis bullosa and the risk of life-reatening cancers : the National EB Registry experience, 1986-2006. J Am Acad Dermatol 2009 ; 60(2) : 203-211.
- Fine JD, et al. : La classification de l’épidermolyse bulleuse héréditaire (EB) : Rapport de la troisième réunion de consensus international sur le diagnostic et la classification de l’EB. J Am Acad Dermatol 2008 ; 58(6) : 931-950.
- Bruckner-Tuderman L, et al. : Progress in Epidermolysis bullosa research : summary of DEBRA International Research Conference 2012. J Invest Dermatol 2013 ; 133(9) : 2121-2126.
- Uitto J, Richard G : Progression de l’épidermolyse bulleuse : classification génétique et implications cliniques. Am J Med Genet C Semin Med Genet 2004 ; 131C(1) : 61-74.
- Kuttner V, et al. : Remodelage global du microenvironnement cellulaire dû à la perte de collagène VII. Mol Syst Biol 2013 ; 9 : 657.
- Odorisio T, et al : Les jumeaux monozygotes discordants pour le phénotype de l’épidermolyse bulleuse récessive mettent en évidence le rôle de la signalisation du TGF-bêta dans la modification de la sévérité de la maladie. Hum Mol Genet 2014 ; 23(15) : 3907-3922.
- Pulkkinen L, et al. : Nouvelles mutations ITGB4 dans les variantes léthales et non léthales de l’épidermolyse bulleuse avec atrésie pylorique : missense versus nonsense. Am J Hum Genet 1998 ; 63(5) : 1376-1387.
- Fine JD, et al : Epidermolyse bulleuse interne : mise à jour des recommandations sur le diagnostic et la classification. J Am Acad Dermatol 2014 ; 70(6) : 1103-1126.
- He Y, et al : Des mutations monoalléliques dans le codon d’initiation de la traduction de KLHL24 provoquent une fragilité de la peau. Am J Hum Genet 2016 ; 99(6) : 1395-1404.
- Lee JYW, et al : Mutations dans KLHL24 Ajouter à l’hétérogénéité moléculaire de l’épidermolyse bulleuse simplexe. J Invest Dermatol 2017 ; 137(6) : 1378-1380.
- Lin Z, et al : Des mutations stabilisantes de la ligase ubiquitine KLHL24 provoquent la perte de la kératine 14 et la fragilité de la peau humaine. Nat Genet 2016 ; 48(12) : 1508-1516.
- Vahidnezhad H, et al : La mutation récessive de la tétraspanine CD151 provoque une épidermolyse bulleuse semblable au syndrome de Kindler avec des manifestations multi-systémiques, y compris une néphropathie. Matrix Biol 2018 ; 66 : 22-33.
- Salo AM, et al : Un trouble des tissus connectifs causé par des mutations du gène de la lysyl hydroxylase 3. Am J Hum Genet 2008 ; 83(4) : 495-503.
- Nischler E, et al : Diagnostic pitfalls in newborns and babies with blisters and erosions. Dermatol Res Pract 2009 ; 2009 : 320403.
- El Hachem M, et al : Multicentre consensus recommendations for skin care inherited epidermolysis bullosa. Orphanet J Rare Dis 2014 ; 9 : 76.
- Dănescu S, et al : Corrélation entre la sévérité de la maladie et la qualité de vie chez les patients atteints d’épidermolyse bulleuse. J Eur Acad Dermatol Venereol 2018. doi : 10.1111/jdv.15371. [Epub ahead of print]
- Mavilio F, et al : Correction de l’épidermolyse bulleuse jonctionnelle par transplantation de cellules souches épidermiques génétiquement modifiées. Nat Med 2006 ; 12(12) : 1397-1402.
- Hirsch T, et al : Régénération de l’épiderme humain entier à l’aide de cellules souches transgéniques. Nature, 2017 ; 551(7680) : 327-332.
- Siprashvili Z, et al. : Sécurité et résultats des plaies suite à des greffons épidermiques autologues génétiquement corrigés chez des patients atteints d’épidermolyse bulleuse consécutive à une dystrophie. JAMA 2016 ; 316(17) : 1808-1817.
- Rosa J, et al : Systèmes actuels de délivrance de siRNA non viraux comme traitement prometteur des maladies de la peau. Curr Pharm Des 2018 ; 24(23) : 2644-2663.
- McBride JD, et al. : Dual mechanism of type VII collagen transfer by bone marrow mesenchymal stem cell extracellular vesicles to recessive dystrophic epidermolysis bullosa fibroblasts. Biochimie 2018 ; 155 : 50-58.
- Hainzl S, et al : Édition de COL7A1 via CRISPR/Cas9 dans la dystrophie récessive de l’épidermolyse bulleuse. Mol Ther 2017 ; 25(11) : 2573-2584.
- March OP, Reichelt J, Koller U : Édition de gènes pour les maladies de la peau : les nucléases de conception comme outils pour la thérapie génétique des troubles de la fragilité de la peau. Exp Physiol 2018 ; 103(4) : 449-455.
- Gostynski A, Pasmooij AM, Jonkman MF : Successful therapeutic transplantation of revertant skin in epidermolysis bullosa. J Am Acad Dermatol 2014 ; 70(1) : 98-101.
- van den Akker PC, et al : Une “cellule révertante tardive-finale” explique la fréquence élevée du mosaïcisme révert dans l’épidermolyse bulleuse. PLoS One 2018 ; 13(2) : p. e0192994.
- Ebens CL, et al : La greffe de moelle osseuse avec du cyclophosphamide post-transplantation pour l’épidermolyse bulleuse dystrophique récessive élargit le pool de donneurs associés et permet la tolérance des greffons cellulaires non hématopoïétiques. Br J Dermatol 2019. doi : 10.1111/bjd.17858. [Epub ahead of print]
- Vanden Oever M, et al : Inside out : médecine régénérative pour l’épidermolyse bulleuse dystrophique récessive. Pediatr Res 2018 ; 83(1-2) : 318-324.
- Ganier C, et al : Injection intradermique de cellules mésenchymateuses de la moelle osseuse corrigeant la dystrophie récessive de l’épidermolyse bulleuse dans un modèle de xénogreffe. J Invest Dermatol 2018 ; 138(11) : 2483-2486.
- Petrof G, et al : Fibroblast cell therapy enhances initial healing in recessive dystrophic epidermolysis bullosa wounds : results of a randomized, vehicle-controlled trial. Br J Dermatol 2013 ; 169(5) : 1025-1033.
- Venugopal SS, et al : A phase II randomised vehicle-controlled trial of intradermal allogeneic fibroblasts for recessive dystrophic epidermolysis bullosa. J Am Acad Dermatol 2013 ; 69(6) : 898-908.e7. doi : 10.1016/j.jaad.2013.08.014
- Itoh M, Kiuru M, Cairo MS, Christiano AM. : Génération de kératinocytes à partir de cellules souches pluripotentes induites par l’épidermolyse bulleuse normale et dystrophique récessive. Proc Natl Acad Sci U S A 2011 ; 108(21) : 8797-8802.
- Nakayama C, et al. : Le développement de cellules mésenchymateuses dérivées de cellules souches pluripotentes induites/cellules stromales à partir de kératinocytes épidermiques humains normaux et RDEB. J Dermatol Sci 2018 ; 91(3) : 301-310.
- South AP, Uitto J : Type VII Collagen Replacement Therapy in Recessive Dystrophic Epidermolysis Bullosa-How Much, How Often ? J Invest Dermatol 2016 ; 136(6) : 1079-1081.
- NIH : Topical Bercolagene Telserpavec (KB103) Gene Therapy to Restore Functional Collagen VII for the Treatment of Dystrophic Epidermolysis Bullosa (GEM-1), https://clinicaltrials.gov/ct2/show/NCT03536143, dernière consultation 25.03.2019.
- Lincoln V, et al : Gentamicin induces LAMB3 nonsense mutation readthrough and restores functional laminin 332 in junctional epidermolysis bullosa. Proc Natl Acad Sci U S A, 2018 ; 115(28) : E6536-E6545.
- Peking P, et al : A Gene Gun-mediated Nonviral RNA trans-splicing Strategy for Col7a1 Repair. Mol Ther Nucleic Acids 2016. 5:e287. doi : 10.1038/mtna.2016.3.
- Turczynski S, et al : Targeted Exon Skipping Restores Type VII Collagen Expression and Anchoring Fibril Formation in an In Vivo RDEB Model. J Invest Dermatol 2016 ; 136(12) : 2387-2395.
- Guttmann-Gruber C, et al : Low-dose calcipotriol can elicit wound closure, anti-microbial, and anti-neoplastic effects in epidermolysis bullosa keratinocytes. Sci Rep 2018 ; 8(1) : 13430.
- Wally V, et al : Diacerein orphan drug development for epidermolysis bullosa simplex : A phase 2/3 randomized, placebo-controlled, double-blind clinical trial. J Am Acad Dermatol 2018 ; 78(5) : 892-901.e7. doi : 10.1016/j.jaad.2018.01.019.
- Atanasova VS, et al : Identification du rigosertib pour le traitement du carcinome épidermoïde dystrophique récessif associé à la bulle. Clin Cancer Res, 2019. doi : 10.1158/1078–0432.CCR-18–2661
DERMATOLOGIE PRATIQUE 2019 ; 29(2) : 16-20









