Dr Basile Darbellay, MD, MD-PhD (Cabinet Dr Basile Darbellay Orsières) & Prof. Christoph Schlapbach, MD, PhD (Inselspital Berne)
La dermatite atopique se caractérise par des lésions cutanées eczématiformes, de fortes démangeaisons ainsi que par une hétérogénéité importante des signes cliniques et de la sévérité [1]. Pour les patients atteints de DA modérée à sévère, il existe maintenant des immunothérapies systémiques ciblées [2]. Dans cette CME, nous présenterons les études pivotales les plus importantes concernant ces approches thérapeutiques modernes.
La dermatite atopique (DA) est l’affection cutanée inflammatoire chronique la plus fréquente dans le monde industrialisé, avec une prévalence croissante dans les pays à revenu moyen et faible [1, 3]. La maladie peut apparaître à tout âge, mais elle commence toutefois au cours des cinq premières années chez 85 % des personnes atteintes et se manifeste typiquement dès le premier âge[4, 5]. En Suisse, environ 20 % des enfants sont touchés par la DA. Chez les adultes, entre 4 % et 5 % de la population sont concernés [4]. La DA se caractérise par une forte hétérogénéité en termes de signes cliniques, de degré de sévérité et d’évolution. Les symptômes typiques sont les fortes démangeaisons, la sécheresse cutanée globale et la présence de lésions cutanées chroniques ou récidivantes, dont la localisation varie en fonction de l’âge [1].
Les lésions cutanées eczémateuses et les fortes démangeaisons peuvent entraîner un manque de sommeil, un stress psychologique, une stigmatisation et une faible estime de soi chez les patients atteints de DA. Il peut en résulter des dépressions, des troubles anxieux et un risque accru de pensées suicidaires [1]. À cela s’ajoute la limitation grandissante de la capacité de travail lorsque la sévérité de la DA augmente [6, 7]. De manière générale, la DA altère ainsi fortement la qualité de vie des personnes concernées et de leur famille. Cela a été également démontré par une étude épidémiologique suédoise publiée récemment, qui a pu mettre en évidence, à partir de données recueillies dès la naissance, un effet négatif de la DA sur la qualité de vie liée à la santé (HRQoL) des jeunes, et cela jusqu’à l’âge adulte. En outre, les données de l’étude indiquent que les personnes affectées ne demandent que rarement conseil à un médecin malgré une HRQoL limitée, ce qui peut entraîner un retard dans le diagnostic de la DA et donc, un sous-traitement [8].
La DA est associée à diverses comorbidités allergiques, auto-immunes et cardiovasculaires [1]. Les comorbidités allergiques telles que les allergies alimentaires, l’asthme et la rhinite allergique se développent au cours de ce que l’on appelle la marche atopique, souvent de manière progressive après l’apparition de la DA dans l’enfance [9]. Les mécanismes exacts qui sous-tendent la progression de la marche atopique ne sont pas encore connus. On ne sait pas encore clairement si une intervention thérapeutique efficace précoce pourrait arrêter celle-ci, même si certaines études l’indiquent [10].
Comment évaluer la sévérité de la DA?
Le choix du traitement adapté dépend, en première ligne, du degré de sévérité clinique de la maladie. Celui-ci est mesuré à l’aide de différents scores. Ceux-ci évaluent les signes cliniques ainsi que le ressenti subjectif des patients et doivent être utilisés en association [11]. Une sélection des scores importants pertinents est résumée ci-dessous:
- EASI (Eczema Area and Severity Index, indice de sévérité et d’étendue de l’eczéma): Exclusivement destiné à la classification des lésions, n’est pas adapté aux symptômes subjectifs des patients [11]; EASI 0 = pas de symptômes; 0,1 – 5,9 = légère; 6,0 – 22,9 = modérée; 23,0 – 72 = sévère [12]
- SCORAD (Scoring of Atopic Dermatitis, notation de la dermatite atopique) [11]: Comprend l’étendue, l’intensité et les symptômes subjectifs du prurit et de l’insomnie [13]; SCORAD < 25 = légère; 25 – 50 = modérée; > 50 – 103 = sévère [11]
- PO-SCORAD (SCORAD établi par le patient): La sévérité de la DA est évaluée indépendamment du médecin, est corrélé au SCORAD [11, 14]
- POEM (Patient-Oriented Eczema measures for Eczema, mesures de l’eczéma par le patient): utilisé pour mesurer des symptômes subjectifs, pas pour les signes objectifs de la maladie dans les études cliniques [11]
- IGA (Investigator’s Global Assessment, évaluation globale de l’investigateur): évaluation globale réalisée par un médecin traitant [11]; utilise une échelle de 4 à 7 points [15]
- WP-NRS (Worst Pruritus Numerical Rating Scale, échelle d’évaluation numérique du pire prurit): échelle (0 – 10) destinée à évaluer l’intensité de la démangeaison la plus importante chez les patients adultes atteints de DA modérée à sévère [16]
Quel traitement pour quel degré de sévérité? – Le traitement graduel!
À partir de la classification de la DA en légère, modérée et sévère, un programme thérapeutique gradué est recommandé dans les directives de traitement, basées sur le consensus européen de 2018. Les options thérapeutiques vont des corticostéroïdes topiques (CST), ou inhibiteurs de la calcineurine à l’immunosuppression ou immunomodulation systémique en passant par les antiseptiques et à la photothérapie ou les «wet wrapping» [11]. Il n’existe aucune directive spécifique à la Suisse, mais la directive S2k allemande a été actualisée en 2021 et inclut maintenant, dans le programme thérapeutique gradué pour le traitement de la DA modérée à sévère, l’utilisation d’un immunomodulateur, le dupilumab (DUPI), un anticorps monoclonal qui est actuellement la première option thérapeutique biologique en Suisse [17, 18].
Dans les directives européennes et allemandes en vigueur, l’utilisation de crèmes hydratantes et d’huiles de bain et le fait d’éviter certains allergènes reconnus cliniquement constituent le traitement de base recommandé (Figure 1, stade 1). Des programmes d’explication correspondants pour reconnaître et éviter les facteurs déclencheurs complètent ces mesures [4, 11]. En cas de DA modérée (SCORAD < 25, eczéma intermittent, stade 2), un traitement réactif à base de CST (classe II-III) plutôt peu puissants, ou à base d’inhibiteurs de la calcineurine ou d’antiseptiques topiques est recommandé. Une DA modérée/sévère (SCORAD 25 – 50, eczéma récidivant, stade 3) sera traitée de manière proactive par des CST plus puissants (classe II/III) ou le tacrolimus, inhibiteur topique de la calcineurine, ou par photothérapie ou un traitement par «wet wrapping». L’accompagnement psychosomatique et les cures thermales peuvent aussi être envisagés. En cas de DA sévère (SCORAD >50, eczéma persistant, stade 4), une hospitalisation peut s’avérer pertinente et un traitement immunosuppresseur par cyclosporine A, azathioprine, méthotrexate ou mycophénolate mofétil peut être indiqué [11, 17]. En Suisse, parmi ces options thérapeutiques, seule la cyclosporine A est autorisée pour la DA [19]. Chez l’adulte, l’utilisation pendant une courte durée de glucocorticostéroïdes oraux, rarement de PUVA ou d’alitrétinoïne, peut être envisagée [11, 17].

Figure 1: Recommandations thérapeutiques pour la DA, en quatre étapes, en fonction du degré de sévérité de la maladie. *La photothérapie est souvent indiquée à partir du stade 2, toutefois uniquement chez l’adulte et pas chez l’enfant; **Le traitement de première ligne est composé de CST, en cas d’intolérance ou en l’absence d’efficacité et sur des zones particulières (visage, zones intertrigineuses, zones génitales, cuir chevelu chez l’enfant), les inhibiteurs topiques de la calcineurine sont utilisés; ***L’utilisation de médicaments pour soulager les démangeaisons ou d’antiseptiques peut être envisagée en complément. Toutes les options thérapeutiques issues des recommandations ne sont pas présentées. Adapté de [11, 17].
Ces lignes directrices n’étant pas à jour des traitements nouvellement disponibles, l’attitude clinique en Suisse est régie par des directives assécurologiques. En cas d’échec des traitements de première ligne (divers topiques) et de deuxième ligne (photothérapie, immunosuppresseurs systémiques dont le seul autorisé en Suisse pour la DA est la cyclosporine) après au moins un mois en raison de contre-indications ou d’effets secondaires, et si la DA est sévère (SCORAD > 50, EASI > 21, IGA 4, DLQI > 10), les traitements de troisième ligne peuvent être utilisés en cas de DA sévère (SCORAD > 50, EASI > 21, IGA 4, DLQI > 10). Les traitements de troisième ligne de la DA comprennent les anticorps monoclonaux dupilumab et tralokinumab ainsi que les inhibiteurs des JAK abrocitinib, baricitinib et upadacitinib.
Besoins non couverts
Les thérapies systémiques traditionnelles ont souvent une efficacité limitée dans la DA modérée à sévère ou doivent être interrompues en raison d’effets secondaires. De plus, l’utilisation à long terme de corticostéroïdes topiques peut être associée à une série d’effets secondaires locaux et systémiques [20]. Il n’est pas rare que cela entraîne une «peur des corticostéroïdes» chez les patients, ce qui se traduit par une mauvaise adhésion au traitement et une diminution du contrôle de la maladie [21]. Dans une étude d’observation portant sur plus de 800 patients adultes atteints de DA modérée à sévère, sous traitement systémique conventionnel, 81 % des patients ont présenté au moins une poussée de la maladie au cours du mois précédant le sondage, 58 % ont rapporté des symptômes modérés à sévères au cours de la dernière semaine et un prurit persistant et des troubles du sommeil étaient également répandus. De plus, on a pu observer une forte diminution de la qualité de vie ainsi que de la productivité au travail [22]. Ces résultats mettent en évidence le besoin non satisfait de nouvelles options thérapeutiques dans la DA (ce que l’on appelle les «unmet needs») [7, 22]. Il semble également judicieux d’adapter les directives de traitement en tenant compte des options thérapeutiques nouvelles.
Options thérapeutiques ciblées pour le traitement de DA modérée à sévère
Grâce à l’amélioration de la compréhension de la physiopathologie (encadré 1) des affections cutanées inflammatoires chroniques, des immunothérapies ciblées ont pu être développées au cours des dernières années [2]. En Suisse, les agents biologiques DUPI et tralokinumab (TRALO) peuvent ainsi être utilisés depuis avril 2019 et février 2022 respectivement, pour le traitement de la dermatite atopique modérée à sévère, lorsqu’un traitement avec des médicaments topiques soumis à prescription n’a pas permis le contrôle adéquat de la maladie ou lorsqu’il est contre-indiqué [18, 23]. Les options thérapeutiques les plus récentes dans le paysage du traitement de la DA modérée à sévère sont représentées depuis 2021 par les inhibiteurs de JAK (JAKi) à prendre par voie orale, l’upadacitinib (UPA), le baricitinib (BARI) et l’abrocitinib (ABRO) autorisé également en Suisse depuis 2022 [23-25]. Le DUPI est recommandé dans l’actualisation de 2021 des directives S2k et il fait l’objet d’un fort consensus pour les patients atteints de DA modérée à sévère, mais les autres thérapies n’y sont pas encore mentionnées [17].L’intégration des recommandations sur l’utilisation de JAKi dans la DA est attendue dans les prochaines mises à jour des directives de traitement. Les études les plus importantes dans le cadre de l’autorisation de mise sur le marché – et surtout les résultats concernant la dose thérapeutique – de ces trois traitements systémiques modernes sont présentés ci-après. L’accent sera d’abord mis sur les approches thérapeutiques les plus récentes pour la DA basées sur l’inhibition des molécules de signalisation JAK. L’accent sera d’abord mis sur les approches thérapeutiques les plus récentes pour la DA fondées sur l’inhibition des molécules de signalisation JAK. Nous aborderons ensuite les agents biologiques qui bloquent la cascade inflammatoire en se liant à certaines cytokines ou à des récepteurs de cytokines [2].
—
Début encadré 1
Physiopathologie de la DA
La physiopathologie de la DA est complexe et pas encore comprise dans tous ses aspects. En principe, on estime que la maladie est marquée par l’interaction complexe de trois facteurs: un défaut de la fonction barrière de la peau, une altération du microbiome cutané et le dérèglement de la réponse immunitaire Th2 [2]. De plus, des facteurs génétiques et environnementaux jouent un rôle. Les facteurs de risque suivants sont connus pour la DA [1]:
- Des antécédents familiaux positifs de maladies atopiques, en particulier de la DA: La composante héréditaire de la DA a été estimée jusqu’à 75 % dans les études de jumeaux.
- Niveau socio-économique élevé et environnement urbain: Il existe une corrélation entre la DA et le niveau socio-économique, ainsi qu’une disparité ville-campagne dans la prévalence de la DA.
- Origines ethniques: Les études montrent une prévalence supérieure de la DA chez les afro-américains (17 %) par rapport aux caucasiens (11 %).
Les variantes du gène FLG codant la protéine filaggrine sont le plus grand facteur de risque génétique connu pour l’apparition de la DA. La filaggrine est produite par les kératinocytes lors du processus de kératinisation et elle joue un rôle important dans le maintien de la barrière cutanée. Environ 30 % des patients présentent de telles mutations FLG [1]. Les facteurs environnementaux peuvent également avoir une influence négative sur la fonction de barrière de la peau [2, 26]. La diminution de la fonction de barrière de la peau augmente la pénétration d’antigènes et d’irritants, tels que les allergènes de l’air ou les toxines environnementales. Cela conduit à l’activation d’une réponse immunitaire fondée sur Th2, dans laquelle les cytokines IL-4, IL-5, et IL-13 sont sécrétées et la production d’IgE dans les cellules B est stimulée. Les cytokines se lient à des récepteurs de cytokine et activent ainsi la cascade de signalisation JAK/STAT dans les cellules hématopoïétiques et non hématopoïétiques. La liaison de l’IL-4 et de l’IL-13 à leurs récepteurs sur les neurones sensoriels ainsi que les effets médiés par les IgE sont associés aux démangeaisons typiques de la DA [2]. Les kératinocytes activés dans la peau sécrètent en outre des signaux pro-inflammatoires tels que l’IL-33 ou la lymphopoïétine stromale thymique (TSLP) qui contribuent à la réduction de la fonction barrière de la peau et stimulent le recrutement d’autres cellules immunitaires de type 2. [1, 2]. Le fait de se gratter aggrave la destruction de la barrière cutanée et favorise ainsi la pénétration d’allergènes et d’autres substances irritantes, ce qui péjore encore la réaction immunitaire [1].
La peau des patients atteints de DA est en outre davantage colonisée par Staphylococcus aureus que celle des personnes en bonne santé [2, 27]. La question se pose d’un rôle éventuel de la proliférationcutanée de S. aureus à la réduction de la fonction barrière de la peau et à l’apparition de l’eczéma ou si cette prolifération est une simple conséquence secondaire de ces mêmes phénomènes. Les études réalisées sur l’animal indiquent une réaction inflammatoire eczématiforme plus importante en cas de sur-représentation de S. aureus dans le microbiome [27]. Simultanément, les cytokines sécrétées en cas de DA réduisent la fonction barrière de la peau et augmentent ainsi la colonisation par S. aureus [2].
Fin encadré 1
—
Inhibition spécifique de la chaîne de transduction du signal JAK
Des facteurs extra-cellulaires tels que les cytokines peuvent diriger l’expression génétique, et ainsi réguler la croissance et la différenciation cellulaires, par l’intermédiaire de la voie de signalisation des JAK. Il existe quatre molécules de signal JAK chez l’homme, JAK1, JAK2, JAK3 et TYK2, qui se lient à différents récepteurs et jouent ainsi différents rôles physiologiques [28]. La transduction du signal, avec la participation des JAK1 après l’activation des récepteurs des cytokines de type I ou II joue un rôle important dans le dérèglement immunitaire en cas de DA. Les JAKi UPA, BARI et ABRO agissent sur ce mécanisme [2].
- Upadacitinib (Rinvoq®)
UPA (Rinvoq®) est un JAKi administré par voie orale qui agit spécifiquement sur les JAK1 et moins sur les JAK2, les JAK3 ou la TYK2. La substance active peut être utilisée à la dose de 15 mg une fois par jour depuis janvier 2020 dans la polyarthrite rhumatoïde et depuis mars 2021 dans celui de la spondylarthrite axiale et de l’arthrite psoriasique, et elle est également autorisée depuis novembre 2021 pour le traitement des adultes atteints de DA modérée à sévère, lorsque le traitement par les médicaments topiques conventionnels ne permet pas un contrôle adéquat de la maladie ou s’ils ne peuvent pas être utilisés [24].
Dans l’étude de phase III, randomisée, en double aveugle, avec contrôle placebo AD Up, l’UPA a été étudié en association aux CST chez les patients atteints de DA modérée à sévère. Les 901 patients au total, âgés de 12 à 75 ans, ont reçu une fois par jour par voie orale, soit UPA 15 mg, soit UPA 30 mg, soit un placebo, toujours accompagné de CST; les critères d’évaluation principaux de l’étude étaient la réponse EASI 75* et une amélioration des valeurs vIGA-DA** après 16 semaines [29]. Avec UPA 15 mg une fois par jour + CST, 39,6 % (IC à 95 %: 34,1-45,2) des patients ont atteint un score vIGA de 0 ou 1, contre 10,9 % (IC à 95 %: 7,4-14,4; p < 0,0001) sous placebo + CST. La réponse EASI 75 sous UPA 15 mg +CST était, avec 64,6 % (IC à 95 %: 59,1-70,0), significativement plus élevée que dans le groupe placebo (26,4 %; IC à 95 %: 21,5–31,4; p < 0,0001). De plus, une rapidité de la réponse au traitement a été observée avec UPA + CST: dès 2 semaines, un nombre significativement plus élevé de patients avait atteint une réponse EASI 75 sous traitement par UPA (p < 0,0001) ou une amélioration vIGA-DA (p < 0,01) que dans le groupe placebo [29]. Dans le cadre de l’observation à long terme de cette étude, 50,8 % des patients sous UPA 15 mg + CST avaient aussi une réponse EASI 75 après 52 semaines et 33,5 % des patients avaient un score vIGA-DA de 0 ou 1 [30].
Dans les études de phase III, randomisées, en double aveugle, avec contrôle placebo MEASURE Up-1 et MEASURE UP-2, l’efficacité et la tolérance d’UPA 15 mg ou 30 mg administré par voie orale, en monothérapie ont été étudiées. Ici aussi, la réponse EASI 75 et l’amélioration du score vIGA-DA après 16 semaines constituaient les critères principaux d’évaluation. Dans les deux études, avec un total de 1683 patients, ces critères d’évaluation ont été atteints. Plus de 40 % des patients, 53 % dans MEASURE Up-1 et 42 % dans MEASURE Up-2 ont en outre atteint une réponse EASI 90 sous UPA 15 mg et respectivement 52 % et 42 % des patients, un soulagement net des démangeaisons, soit une amélioration d’au moins 4 points sur l’EVA du pire prurit. Dans les groupes placebo, seul un patient sur dix environ a atteint ces deux critères [20]. Dans une analyse intégrée des deux études, après 52 semaines, 81 % des patients sous UPA 15 mg ont présenté une réponse EASI 75, 62 % une réponse EASI 90 et 65 % une nette amélioration des démangeaisons [31]. Dans les études MEASURE Up-1 et MEASURE Up-2, une réponse rapide à l’UPA a également été observée : Après deux jours, un nombre de patients significativement plus important signalait un soulagement net des démangeaisons sous UPA que sous placebo (p < 0,0001)[20].
L’efficacité et la sécurité de l’UPA et du DUPI ont été comparées directement dans l’étude randomisée de phase III HEADS UP incluant 692 patients adultes atteints de DA modérée à sévère pour lesquels un traitement systémique était envisagé. Pour cela, les patients ont pris UPA à la dose de 30 mg par jour, non autorisée en Suisse, alors que le DUPI était administré en injections sous-cutanées avec une dose initiale de 600 mg suivie de 300 mg toutes les deux semaines. Dès la première semaine, un nombre significativement plus élevé de patients présentait une amélioration cliniquement significative des démangeaisons dans le groupe UPA par rapport au groupe DUPI, ce qui souligne la réponse rapide à UPA. Après 16 semaines, un nombre significativement plus élevé de patients sous UPA avait une réponse EASI 75 par rapport au DUPI (71 % vs 61 % ; p = 0,006), ce qui a permis d’atteindre le critère d’évaluation principal de l’étude. Pour les réponses EASI 90 et EASI 100 ainsi que pour l’amélioration des démangeaisons, les résultats étaient significativement meilleurs avec UPA qu’avec DUPI. Les effets indésirables les plus fréquents au cours des 16 premières semaines de traitement ont été l’acné sous UPA (16 % vs 3 % sous DUPI) et la conjonctivite sous DUPI (8 % vs 1 % sous UPA). Des effets indésirables graves associés au traitement ayant entraîné l’arrêt de l’étude sont apparus chez 3 % des patients sous UPA et chez 1 % des patients sous DUPI [32]. La réponse à long terme, sur plus de 4 mois, à l’UPA et au DUPI peut être considérée comme comparable au vu des données actuellement disponibles.
Le profil de sécurité de l’UPA a été largement caractérisé dans 4 indications. Comparé aux études cliniques dans l’indication de la polyarthrite rhumatoïde, de la spondylarthrite ankylosante et de l’arthrite psoriasique, l’acné est survenue plus souvent dans les études sur la DA, toutefois dans la plupart des cas, l’évolution a été légère [20]. L’analyse intégrée sur 52 semaines de MEASURE Up 1 et 2, avec un total de 797 patients et 953,3 patients-années sous UPA 15 mg a donné un taux d’acné de 13,1 pour 100 patients-années. La fréquence d’apparition du virus de l’herpès zoster était de 3,6 pour 100 patients-années dans cette analyse. Les malignités sont apparues extrêmement rarement, avec un taux de 0,2 pour 100 patients-années. Les événements cardiovasculaires graves (MACE) et les thromboembolies veineuses (TEV) ont respectivement été observés chez un patient sous UPA 15 mg, qui présentait au moins un facteur de risque cardiovasculaire [31].
Le BARI (Olumiant®) est un inhibiteur de JAKi qui présente une inhibition sélective des molécules du signal JAK1 et JAK2. Ce principe actif est utilisé dans le traitement de la polyarthrite et il est désormais également autorisé pour le traitement de patients adultes atteints de DA modérée à sévère, lorsqu’une thérapie par des médicaments topiques ne permet pas un contrôle adéquat de la maladie ou n’est pas recommandée [25]. Sur la base des études pivotales BREEZE-AD1, 2, 3, 4 et 7, le BARI est utilisé sous la forme d’un comprimé à 4 mg une fois par jour ou sous la forme d’un comprimé à 2 mg deux fois par jour. Si aucun succès thérapeutique n’est visible après 8 semaines de traitement, celui-ci doit être interrompu. Les patients ayant obtenu un contrôle durable de l’activité de la maladie avec 4 mg une fois par jour peuvent passer à une dose d’entretien de BARI 2 mg une fois par jour [22].
Les études de phase III randomisées, en double aveugle, avec contrôle placebo BREEZE-AD1, BREEZE-AD2 et BREEZE-AD7 ont évalué l’efficacité et la tolérance de BARI 1 mg, 2 mg et 4 mg administré une fois par jour par voie orale pendant 16 semaines chez un total de 1568 patients atteints de DA modérée à sévère et ayant une réponse insuffisante ou une intolérance aux médicaments topiques [33-35]. Dans les études BREEZE AD-1 et AD-2, le BARI a été utilisé une fois par jour en monothérapie. Le critère d’évaluation principal était le score IGA 0/1 et une amélioration d’au moins 2 points sur l’échelle IGA. Après 16 semaines, 17 % des patients de BREEZE-AD1 et 14 % des patients de BREEZE-AD2 sous BARI 4 mg ont présenté une amélioration du score IGA, contre 5 % (p<0,001 dans les deux cas) dans chaque bras placebo. Une réponse EASI 75 a été atteinte avec la dose la plus élevée en monothérapie par 25 % des patients de BREEZE-AD1 et par 21 % des patients de BREEZE-AD2. Dans les groupes placebo, 6-9 % des patients seulement y sont parvenus (pour les deux, p ≤ 0,001). Une amélioration des démangeaisons a été observée avec la dose de BARI de 4 mg dès 1 semaine, tout comme l’amélioration du sommeil et de la qualité de vie (p ≤ 0,05 pour toutes les comparaisons) [33]. Dans l’étude BREEZE-AD7, une dose de BARI 4 mg a ensuite aussi été testée en association aux CST et a permis une amélioration du score IGA chez 31 % des patients après 16 semaines, comparé à 15 % dans le groupe placebo (p = 0,004) [35]
Dans l’étude d’extension BREEZE-AD3, des patients des études BREEZE-AD1 et AD2 qui avaient présenté une réponse complète ou partielle au traitement ont reçu un traitement par BARI jusqu’à 68 semaines [36]. Le critère d’évaluation principal était une amélioration du score IGA après 16, 36 et 52 semaines de l’étude d’extension. La réponse IGA est restée stable pendant toute la durée du traitement sous BARI 4 mg. Au début de l’étude BREEZE-AD3, 46 % des patients présentaient un score IGA de 0 ou 1; à la fin de l’étude, soit après 68 semaines de traitement au total, ils étaient 47 %. La réduction des démangeaisons est également restée stable pendant la durée du traitement par BARI 4 mg. Pour la réponse EASI 75, une légère réduction dans la durée a été observée: 70 % des patients présentaient une réponse EASI 75 au début de BREEZE-AD3 avec BARI 4 mg, alors qu’ils étaient encore 56 % après 68 semaines de traitement [36].
Dans l’étude de phase III, randomisée, en double aveugle, avec contrôle placebo BREEZE-AD4, avec 462 participants, l’association BARI + CST a également été testée chez des patients atteints de DA modérée à sévère, présentant une intolérance ou une contre-indication au traitement à la cyclosporine par voie orale. Dans cette étude, 13 % des patients ayant reçu un traitement par BARI 4 mg + CST et 13 % des patients sous placebo + CST ont atteint un score IGA de 0 ou 1 et une amélioration d’au moins 2 points sur l’échelle IGA (p ≤ 0,05) après 16 semaines. 28 % des patients sous BARI 4 mg + CST ont atteint une réponse EASI 75 et ils étaient 15 % sous placebo + CST (p ≤ 0,05) [25, 37].
Selon une analyse de sécurité intégrée à long terme de huit études cliniques incluant un total de 2531 patients et 2247 patients-années sous BARI, les infections opportunistes graves et les conjonctivites sont rarement survenues, et avec une fréquence comparable pour les doses de 2 mg et 4 mg et sous placebo. L’incidence du virus de l’herpès zoster était de 2,3/100 000 pour les deux dosages combinés, celle de l’herpès simplex était de 10,3. Avec une incidence de 0,2, les malignités ont été rares, comme les MACE et les TEV qui n’ont été observés que chez deux patients respectivement[38].
- Abrocitinib (Cibinqo®)
Le JAKi oral ABRO (Cibinqo®) inhibe principalement JAK1 et peut être utilisé à la posologie de 100 mg une fois par jour depuis avril 2022 pour le traitement de patients adultes atteints de DA modérée à sévère, lorsqu’une thérapie par des médicaments topiques ne permet pas un contrôle adéquat de la maladie ou n’est pas recommandée. Si aucun succès thérapeutique n’est visible après 12 semaines de traitement, la prise du médicament doit être interrompue [39].
Les études de phase III, randomisées, en double aveugle, avec contrôle placebo JADE MONO-1, MONO-2 et COMPARE ont évalué l’efficacité et la sécurité d’ABRO en monothérapie par rapport à un placebo ou au DUPI[40-42]. L’étude JADE MONO-1 a porté sur 387 patients âgés de 12 ans et plus, atteints de DA modérée à sévère qui ont reçu une fois par jour pendant 12 semaines un placebo, 100 mg ou 200 mg d’ABRO. Les critères d’évaluation co-primaires étaient un score IGA 0/1 ou une amélioration sur l’échelle IGA d’au moins 2 points ainsi qu’une réponse EASI 75 à 12 semaines. Un nombre significativement plus élevé de patients dans le groupe ABRO 100 mg (24 %) que dans le groupe placebo (8 %) ont atteint une réponse IGA (p = 0,0037). 40 % des patients sous ABRO 100 mg et 12 % des patients sous placebo (p < 0,0001) ont obtenu une réponse EASI 75. Des effets indésirables graves ont été rapportés chez 3 % des patients recevant 100 mg d’ABRO et 4 % des patients recevant le placebo[40]. L’étude JADE MONO-2, dont le plan d’étude est identique à celui de JADE MONO-1, a porté sur 391 patients. 28 % des patients sous ABRO 100 mg ont obtenu ici une amélioration du score IGA et 45 % une réponse EASI 75, contre 9 % et 10 % des patients sous placebo (p < 0,001 dans les deux cas). Dans cette étude également, la fréquence des effets indésirables graves était comparable dans le groupe ABRO 100 mg et dans le groupe placebo (3 % contre 1 %). Une réduction du nombre des plaquettes et des valeurs de laboratoire indiquant une thrombocytopénie ont été mises en évidence dans le groupe ABRO 200 mg[41].
Dans l’étude comparative de phase III en double aveugle JADE COMPARE, 838 patients ont été traités une fois par jour pendant 12 semaines avec 100 mg ou 200 mg d’ABRO par voie orale, ont reçu un placebo ou ont reçu 300 mg de DUPI par voie sous-cutanée toutes les 2 semaines après une dose initiale de 600 mg. Les critères d’évaluation primaires d’une amélioration du vIGA et d’une réponse EASI 75, comme dans les études JADE MONO-1 et -2, ont été atteints ici à 12 semaines par 37 %, 37 % et 14 % ou 59 %, 58 % et 27 % des patients du groupe ABRO 100 mg, du groupe DUPI et du groupe placebo (p < 0,001 pour ABRO vs placebo pour l’IGA et l’EASI 75). Des nausées sont survenues chez 4,2 % des patients et de l’acné chez 2,9 % des patients sous ABRO 100 mg[42].
Une analyse groupée des études JADE MONO-1 et MONO-2 et de l’étude de phase IIb a également examiné les résultats rapportés par les patients (PRO) avec le traitement ABRO. Les analyses ont porté sur 942 patients. Sous ABRO 100 mg, 43 % des patients ont présenté une amélioration des démangeaisons nocturnes (Amélioration Night Time Itch Scale ≥ 4 points) et 65 % une amélioration d’un point du score PSAAD(Prurit and Symptoms Assessment for AD), contre 13 % et 34 % dans le groupe placebo. L’état mental des patients s’est également amélioré après 12 semaines sous ABRO 100 mg, avec une réduction de 1,7 pour l’anxiété et de 1,3 pour la dépression sur les échelles Hospital Anxiety and Depression Scales (HADS), contre 1,0 et 0,1 dans le groupe placebo[43].
Informations générales pertinentes pour l’utilisation des JAKi:
- Prise sous forme de comprimés, indépendante des repas [24, 25]
- En association aux CST ou en monothérapie [24, 25]
- Ne pas les associer à d’autres immunosuppresseurs puissants comme la cyclosporine, les immunomodulateurs biologiques ou les JAKi [24, 25]
- Ne sont pas recommandés dans les cas suivants: [24, 25]
- Hypersensibilité au principe actif ou à l’un des excipients
- Troubles sévères de la fonction hépatique
- Tuberculose active ou infections graves
- En cas de grossesse et pendant l’allaitement
- Si numération absolue des lymphocytes (ALC) < 500 cellules/mm3
- Si numération absolue des neutrophiles (ANC) < 1000 cellules/mm3
- Si taux d’hémoglobine < 8 g/dl
- Si taux de filtration glomérulaire estimé < 30 ml/min/1,73 m2 (BARI)[25]
- L’UPA n’a pas été étudié chez les patients présentant une insuffisance rénale terminale [24].
- Avant le début du traitement, mettre le statut vaccinal à jour, y compris pour le vaccin contre le virus de l’herpès zoster, conformément aux recommandations vaccinales en vigueur [24, 25]
- Exclusion d’infections chroniques avant le traitement et contrôles sanguins réguliers pendant le traitement [44]
Inhibition ciblée des récepteurs de l’IL-4 et de la molécule de signal de l’IL-13
- Dupilumab (Dupixent®)
Le DUPI (Dupixent®) est un anticorps monoclonal humain qui se lie spécifiquement au récepteur de l’IL-4 type I et de l’IL-4/IL-13 type II et inhibe ainsi la transduction du signal médiée par l’IL-4 et l’IL-13. Le DUPI est utilisé comme traitement adjuvant d’entretien en cas d’asthme sévère et il est également indiqué comme traitement adjuvant en cas de rhinosinusite chronique avec polypose nasale. En Suisse, le principe actif est autorisé depuis 2019 pour le traitement de la DA modérée à sévère chez les adultes et les adolescents de plus de 12 ans, lorsqu’une thérapie par des médicaments topiques soumis à prescription médicale ne permet pas un contrôle adéquat de la maladie ou n’est pas recommandée. Le DUPI est disponible en seringue prête à l’emploi aux doses de 200 mg et 300 mg et a été évalué dans le cadre des trois études pivotales SOLO 1, SOLO 2 et CHRONOS. Selon ces études, le DUPI peut être utilisé en monothérapie ou en association aux CST. Chez les patients qui n’ont pas répondu au traitement après 16 semaines, il convient d’envisager l’arrêt de celui-ci [18].
Dans les études parallèles de phase III, randomisées, en double aveugle, avec contrôle placebo SOLO 1 et SOLO 2, le DUPI a été étudié en monothérapie sur un total de 1379 patients atteints de DA modérée à sévère chez lesquels les médicaments topiques n’avaient pas fonctionné ou n’avaient pas fonctionné suffisamment. Les patients recevaient une dose initiale de 600 mg de DUPI, puis 300 mg de DUPI par semaine (qw) ou toutes les deux semaines (q2w); le critère d’évaluation principal était l’amélioration du score vIGA-DA et le critère d’évaluation secondaire était une réponse EASI 75 après 16 semaines [45, 46]. En Suisse, seul le traitement q2w est autorisé pour le DUPI [18]. Avec celui-ci, 37 % des patients ont atteint une amélioration du score vIGA-DA et 48 % une réponse EASI 75, comme le précise une analyse groupée des deux études. Dans les groupes placebo, seuls 9 % et 13 % des patients y sont parvenus (p < 0,0001) [46]. Le DUPI a en outre présenté une amélioration significative rapide des démangeaisons, après 4 jours seulement, par rapport au bras placebo (p < 0,0001) [46].
Dans l’étude de phase III, randomisée, en double aveugle, avec contrôle placebo CHRONOS, le DUPI a été évalué en association aux CST. Les patients atteints de DA modérée à sévère et présentant une réponse inadéquate aux CST ont été traités pendant 52 semaines par DUPI 300 mg qw ou q2w et recevaient en plus des CST comme indiqué en fonction du degré de la maladie [47]. Les critères d’évaluation co-primaires étaient un score IGA 0/1 et une amélioration du score IGA d’au moins 2 points ainsi qu’une réponse EASI 75. Après 16 semaines, 39 % des patients recevant un traitement par DUPI + CST q2w ont atteint le critère IGA et seulement 12 % des patients du groupe placebo (p < 0,0001). Une réponse EASI 75 a été observée chez 69 % de ces patients et chez 23 % des patients sous placebo (p < 0,0001). L’efficacité s’est montrée stable sur 52 semaines, 36 % des patients recevant un traitement par DUPI + CST q2w avaient atteint le critère IGA à la fin de l’étude et 65 % le critère EASI 75 (p < 0,0001)[47].
Des réactions au site d’injection ainsi que des conjonctivites, des nasopharyngites et des infections des voies respiratoires supérieures étaient des effets indésirables du traitement par DUPI [45, 47]. Dans le groupe placebo + CST de l’étude CHRONOS, les taux d’effets indésirables graves et d’arrêt du traitement en raison des effets indésirables étaient d’ailleurs plus élevés que dans le bras de traitement par DUPI [47].
- Tralokinumab (Adtralza®)
L’anticorps humain TRALO (Adtralza®) se lie spécifiquement à l’IL-13, inhibant ainsi l’activation du complexe récepteur IL-13Rα1/IL-4Rα. Depuis février 2022, le TRALO est autorisé en Suisse pour le traitement des patients adultes atteints de DA modérée à sévère, lorsque le traitement par des médicaments topiques délivrés sur ordonnance ne permet pas un contrôle adéquat de la maladie ou n’est pas recommandé. Une dose initiale de 600 mg de TRALO en injection sous-cutanée est recommandée, puis des injections bihebdomadaires de 300 mg de TRALO. La posologie peut être réduite à une injection sous-cutanée toutes les quatre semaines lorsque le patient a atteint une cicatrisation complète ou presque complète de la peau[23].
Les trois études de phase III randomisées, en double aveugle, avec contrôle placebo ECZTRA 1, ECZTRA 2 et ECZTRA 3 ont évalué l’efficacité et la sécurité du TRALO chez un total de 1976 adultes atteints de DA modérée à sévère[23, 48, 49]. Dans l’étude ECZTRA 1 portant sur 802 patients et l’étude ECZTRA 2 portant sur 794 patients, TRALO a été utilisé en monothérapie aux doses décrites ci-dessus et comparé à un placebo. Les critères d’évaluation primaires étaient une amélioration de l’IGA et une réponse EASI 75 après 16 semaines. Les patients qui ont atteint au moins l’un de ces critères d’évaluation primaires ont été randomisés après 16 semaines dans une deuxième phase de l’étude pour recevoir une dose de 300 mg de TRALO ou de placebo toutes les 2 ou 4 semaines pendant 36 semaines supplémentaires. Après 16 semaines, 16 % et 22 % des patients sous TRALO et 7 % et 11 % des patients sous placebo dans ECZTRA 1 (IC à 95 %: 4,1-13,1; p=0,002) et ECZTRA 2 (IC à 95 %: 5,8-16,4; p<0,001) avaient atteint une amélioration IGA. Une réponse EASI 75 a été observée chez 25 % et 33 % des patients sous TRALO contre 13 % et 11 % du groupe placebo dans le cadre d’ECZTRA 1 (IC à 95 %: 6,5-17,7; p<0,001) et étude ECZTRA 2 (IC à 95 %: 15,8–27,3; p<0,001). Les PRO ont mis en évidence une amélioration précoce des démangeaisons, du sommeil et de la qualité de vie des patients, et la réponse au traitement par TRALO s’est maintenue jusqu’à 52 semaines chez la majorité des patients. Le traitement a été bien toléré jusqu’à 52 semaines, avec des effets indésirables dans 76 % et 62 % des cas du groupe TRALO et 77 % et 66 % des cas du groupe placebo dans les deux études[48].
L’étude ECZTRA 3 a évalué l’efficacité et la sécurité du TRALO en combinaison avec un TCS par rapport à l’association placebo + TCS. La conception de l’étude, d’une durée totale de 32 semaines, était similaire à celle des deux premières études de phase III, à la différence près que la deuxième phase, portant sur des patients ayant répondu au traitement, n’a duré que 16 semaines supplémentaires. Avec une injection bihebdomadaire de 300 mg de TRALO + TCS à la demande, 39 % des patients ont obtenu une amélioration de l’IGA et 56 % des patients ont obtenu une réponse EASI 75 après 16 semaines. Dans le groupe placebo + TCS, 26 % y sont parvenus (IC à 95 %: 2,9-21,9; p=0,015) et 36 % (IC à 95 %; 9,8-30,6; p<0,001) des patients. Après 32 semaines, 90 % et 93 % des patients ayant reçu des injections bihebdomadaires de TRALO, ainsi que 78 % et 91 % des patients ayant reçu des injections quadrihebdomadaires de TRALO ont maintenu l’amélioration de l’IGA et de la réponse EASI 75. L’incidence des effets indésirables était également comparable entre les groupes. Les infections des voies respiratoires supérieures, la conjonctivite, les maux de tête et les réactions au point d’injection ont été des effets indésirables fréquents, apparus plus souvent sous TRALO que sous placebo[49].
Informations pertinentes pour l’utilisation du DUPI et du TRALO[18, 23]:
- S’utilise en injection sous-cutanée
- En association aux CST ou en monothérapie
- N’est pas recommandé dans les cas suivants:
- Hypersensibilité au principe actif ou à l’un des excipients
- Symptômes d’asthme aigu, exacerbations aiguës, bronchospasme aigu ou asthme aigu grave (pour DUPI)
- Helminthiase
- Grossesse
- Pas ou peu étudié chez
- Les patients présentant une insuffisance hépatique
- Les patients atteints d’insuffisance rénale sévère
- Les femmes pendant l’allaitement
- Avant le début du traitement, mettre le statut vaccinal à jour, y compris pour le vaccin contre le virus de l’herpès zoster, conformément aux recommandations vaccinales en vigueur
Traitements de la DA dans le pipeline [50]
Plus de 80 nouveaux traitements pour la DA sont actuellement dans le pipeline. Les modes d’action de ces traitements en cours d’élaboration vont de la modification du microbiome cutané au traitement ciblé de la réaction immunitaire excessive en passant par le rétablissement de la fonction barrière de la peau. Les traitements en cours de développement à partir de la phase II sont énumérés dans le tableau ci-après [50]:
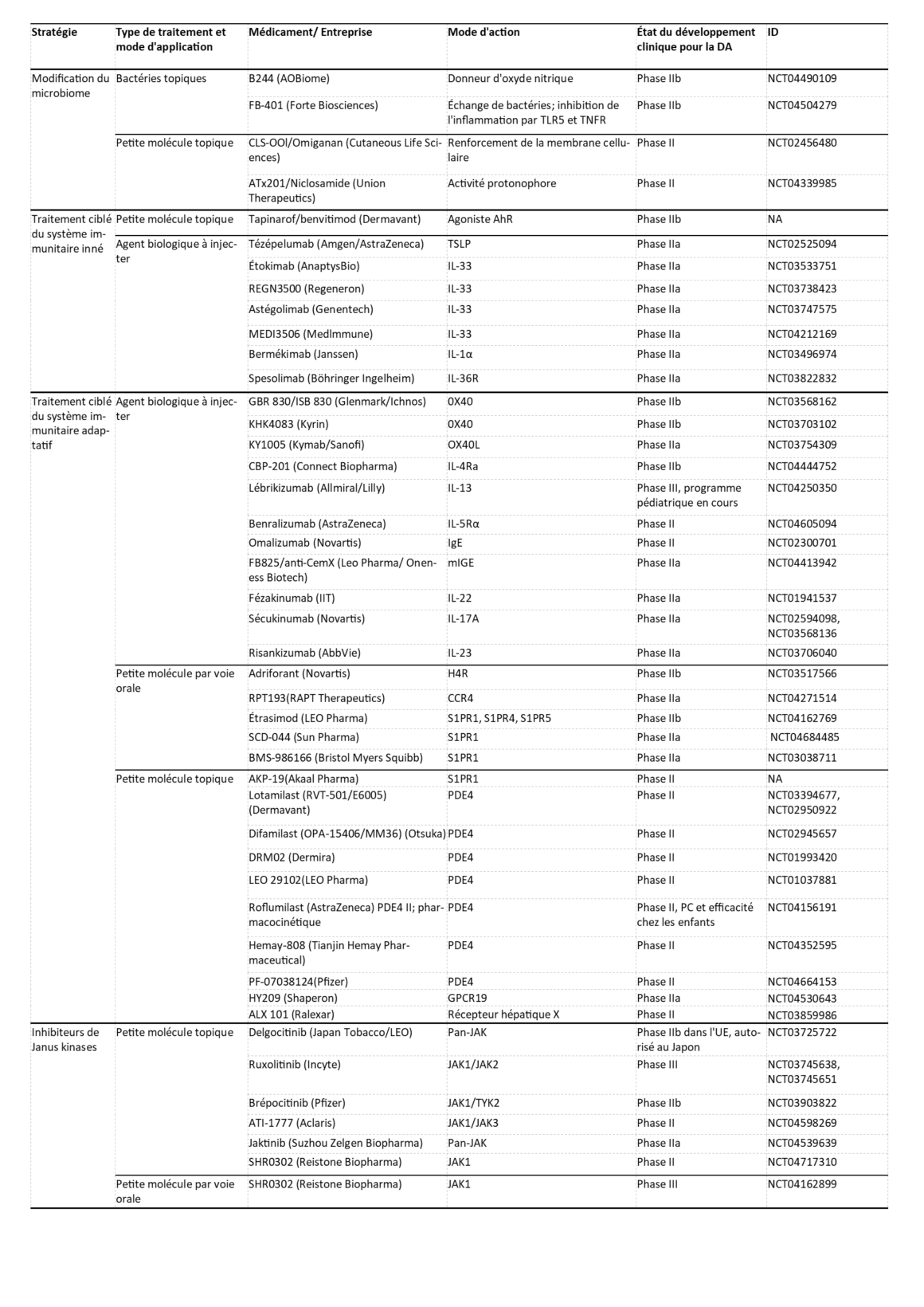
| Légende: AhR, récepteur aryl-hydrocarboné ; CCR4, récepteur C- C des chimiokines 4 ; GPCR19, récepteur 19 couplé aux protéines G ; H4R, récepteur de type 4 de l’histamine ; IL-4Rα, chaîne α du récepteur IL-4 ; IL-5Rα, chaîne α du récepteur IL-5 ; IL-13Rα1, chaîne α1 du récepteur de l’IL-13 ; IL-22R1, récepteur 1 de l’IL-22 ; JAK, Janus kinase ; NA, sans objet ; OX40L, ligand de l’OX40 ; PDE4, phosphodiestérase 4 ; S1PR1, récepteur 1 de la sphingosine 1-phosphate ; TSLP, lymphopoïétine du stroma thymique. |
– La physiopathologie de la DA est une interaction complexe ‘d’une fonction barrière perturbée de la peau, d’une altération du microbiome cutané et du dérèglement de la réponse immunitaire Th2 .
– Les progrès dans la compréhension des bases immunologiques de la DA permettent le développement d’immunothérapies ciblées, parmi lesquelles les JAKi oraux upadacitinib, baricitinib et abrocitinib sont à ce jour autorisés en Suisse, en plus des agents biologiques injectables que sont le dupilumab et le tralokinumab.
– Les immunothérapies montrent une bonne efficacité et un profil de tolérance favorable, également dans les études à long terme.
– L’hétérogénéité de la maladie fait que même avec les immunothérapies, il n’existe pas de « modèle unique » pour le traitement.
*Réponse EASI 75: Réduction de 75 % de la valeur EASI par rapport à la valeur initiale
** Amélioration du score vIGA-DA: Valeur vIGA-DA de 0 ou 1 avec ≥ 2 points d’amélioration par rapport à la valeur initiale
Littérature
1. Langan SM et al. Atopic dermatitis. Lancet, 2020. 396(10247): p. 345-360.
2. Song A et al. Immunopathology and Immunotherapy of Inflammatory Skin Diseases. Immune Netw, 2022. 22(1): p. e7.
3. Weidinger S et al. Atopic dermatitis. Nat Rev Dis Primers, 2018. 4(1): p. 1.
4. Website vom Allergiezentrum Schweiz zur Neurodermitis. Einsehbar auf https://www.aha.ch/allergiezentrum-schweiz/haut/neurodermitis-atopisches-ekzem. Letzter Zugriff: März 2022.
5. Barbarot S et al. Epidemiology of atopic dermatitis in adults: Results from an international survey. Allergy, 2018. 73(6): p. 1284-1293.
6. Barrett A et al. Patient-Reported Outcome Measures in Atopic Dermatitis and Chronic Hand Eczema in Adults. Patient, 2019. 12(5): p. 445-459.
7. Simon D et al. Atopic Dermatitis: Collegium Internationale Allergologicum (CIA) Update 2019. International Archives of Allergy and Immunology, 2019. 178(3): p. 207-218.
8. Lundin S et al. Living with Atopic Dermatitis as a Young Adult in Relation to Health-related Quality of Life and Healthcare Contacts: A Population-based Study. Acta Derm Venereol, 2022.
9. Hill DA et al. The atopic march: Critical evidence and clinical relevance. Ann Allergy Asthma Immunol, 2018. 120(2): p. 131-137.
10. Paller AS et al. The atopic march and atopic multimorbidity: Many trajectories, many pathways. J Allergy Clin Immunol, 2019. 143(1): p. 46-55.
11. Wollenberg A et al. Consensus-based European guidelines for treatment of atopic eczema (atopic dermatitis) in adults and children: part I. J Eur Acad Dermatol Venereol, 2018. 32(5): p. 657-682.
12. Chopra R et al. Severity strata for Eczema Area and Severity Index (EASI), modified EASI, Scoring Atopic Dermatitis (SCORAD), objective SCORAD, Atopic Dermatitis Severity Index and body surface area in adolescents and adults with atopic dermatitis. Br J Dermatol, 2017. 177(5): p. 1316-1321.
13. Severity scoring of atopic dermatitis: the SCORAD index. Consensus Report of the European Task Force on Atopic Dermatitis.Dermatology, 1993. 186(1): p. 23-31.
14. Stalder JF et al. Patient-Oriented SCORAD (PO-SCORAD): a new self-assessment scale in atopic dermatitis validated in Europe.Allergy, 2011. 66(8): p. 1114-21.
15. Futamura M et al. A systematic review of Investigator Global Assessment (IGA) in atopic dermatitis (AD) trials: Many options, no standards. J Am Acad Dermatol, 2016. 74(2): p. 288-94.
16. Yosipovitch G et al. Peak Pruritus Numerical Rating Scale: psychometric validation and responder definition for assessing itch in moderate-to-severe atopic dermatitis. Br J Dermatol, 2019. 181(4): p. 761-769.
17. Werfel T et al. Update “Systemic treatment of atopic dermatitis” of the S2k-guideline on atopic dermatitis. J Dtsch Dermatol Ges, 2021. 19(1): p. 151-168.
18. Aktuelle Fachinformation DUPIXENT® (Dupilumab) auf www.swissmedicinfo.ch.
19. Aktuelle Fachinformation Sandimmun Neoral® (Cyclosporin A) auf www.swissmedicinfo.ch.
20. Guttman-Yassky E et al. Once-daily upadacitinib versus placebo in adolescents and adults with moderate-to-severe atopic dermatitis (Measure Up 1 and Measure Up 2): results from two replicate double-blind, randomised controlled phase 3 trials. Lancet, 2021. 397(10290): p. 2151-2168.
21. Cork MJ et al. Atopic dermatitis epidemiology and unmet need in the United Kingdom. J Dermatolog Treat, 2020. 31(8): p. 801-809.
22. Wei W et al. A real-world study evaluating adeQUacy of Existing Systemic Treatments for patients with moderate-to-severe Atopic Dermatitis (QUEST-AD): Baseline treatment patterns and unmet needs assessment. Ann Allergy Asthma Immunol, 2019. 123(4): p. 381-388.e2.
23. Aktuelle Fachinformation ADTRALZA® (Tralokinumab) auf www.swissmedicinfo.ch.
24. Aktuelle Fachinformation RINVOQ® (Upadacitinib) auf www.swissmedicinfo.ch
25. Aktuelle Fachinformation OLUMIANT® (Baricitinib) auf www.swissmedicinfo.ch.
26. Sandilands A et al. Filaggrin in the frontline: role in skin barrier function and disease. J Cell Sci, 2009. 122(Pt 9): p. 1285-94.
27. Kobayashi T et al. Dysbiosis and Staphylococcus aureus Colonization Drives Inflammation in Atopic Dermatitis. Immunity, 2015. 42(4): p. 756-66.
28. O’Shea JJ et al. The JAK-STAT pathway: impact on human disease and therapeutic intervention. Annu Rev Med, 2015. 66: p. 311-28.
29. Reich K et al. Safety and efficacy of upadacitinib in combination with topical corticosteroids in adolescents and adults with moderate-to-severe atopic dermatitis (AD Up): results from a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet, 2021. 397(10290): p. 2169-2181.
30. Silverberg JI et al. Upadacitinib plus topical corticosteroids in atopic dermatitis: week-52 AD Up study results. J Allergy Clin Immunol, 2021.
31. Simpson EL et al. Efficacy and Safety of Upadacitinib in Patients With Atopic Dermatitis: Results Through Week 52 From Replicate , Phase 3, Randomized, Double Blind, Placebo Controlled Studies: Measure Up 1 and Measure Up 2. Presented at the 2021 Dermatology Education Foundation (DEF) Essential Resource Meeting (DERM2021), August 5-8, 2021, Las Vegas NV, USA.
32. Blauvelt A et al. Efficacy and Safety of Upadacitinib vs Dupilumab in Adults With Moderate-to-Severe Atopic Dermatitis: A Randomized Clinical Trial. JAMA Dermatol, 2021.
33. Simpson EL et al. Baricitinib in patients with moderate-to-severe atopic dermatitis and inadequate response to topical corticosteroids: results from two randomized monotherapy phase III trials. Br J Dermatol, 2020. 183(2): p. 242-255.
34. Thyssen JP et al. Baricitinib Rapidly Improves Skin Pain Resulting in Improved Quality of Life for Patients with Atopic Dermatitis: Analyses from BREEZE-AD1, 2, and 7. Dermatol Ther (Heidelb), 2021. 11(5): p. 1599-1611.
35. Reich K et al. Efficacy and Safety of Baricitinib Combined With Topical Corticosteroids for Treatment of Moderate to Severe Atopic Dermatitis: A Randomized Clinical Trial. JAMA Dermatol, 2020. 156(12): p. 1333-1343.
36. Silverberg JI et al. Long-term Efficacy of Baricitinib in Adults With Moderate to Severe Atopic Dermatitis Who Were Treatment Responders or Partial Responders: An Extension Study of 2 Randomized Clinical Trials. JAMA Dermatol, 2021. 157(6): p. 691-699.
37. A Long-term Study of Baricitinib (LY3009104) With Topical Corticosteroids in Adults With Moderate to Severe Atopic Dermatitis That Are Not Controlled With Cyclosporine or for Those Who Cannot Take Oral Cyclosporine Because it is Not Medically Advisable (BREEZE-AD4). https://clinicaltrials.gov/ct2/show/NCT03428100.
38. Bieber T et al. Pooled safety analysis of baricitinib in adult patients with atopic dermatitis from 8 randomized clinical trials. J Eur Acad Dermatol Venereol, 2021. 35(2): p. 476-485.
39. Aktuelle Fachinformation CIBINQO® (Abrocitinib) auf www.swissmedicinfo.ch.
40. Simpson EL et al. Efficacy and safety of abrocitinib in adults and adolescents with moderate-to-severe atopic dermatitis (JADE MONO-1): a multicentre, double-blind, randomised, placebo-controlled, phase 3 trial. Lancet, 2020. 396(10246): p. 255-266.
41. Silverberg JI et al. Efficacy and Safety of Abrocitinib in Patients With Moderate-to-Severe Atopic Dermatitis: A Randomized Clinical Trial.JAMA Dermatol, 2020. 156(8): p. 863-873.
42. Bieber T et al. Abrocitinib versus Placebo or Dupilumab for Atopic Dermatitis. N Engl J Med, 2021. 384(12): p. 1101-1112.
43. Silverberg JI et al. Impact of Oral Abrocitinib Monotherapy on Patient-Reported Symptoms and Quality of Life in Adolescents and Adults with Moderate-to-Severe Atopic Dermatitis: A Pooled Analysis of Patient-Reported Outcomes. Am J Clin Dermatol, 2021. 22(4): p. 541-554.
44. Schweizerische Gesellschaft für Rheumatologie, Fachinformationen, Behandlungsempfehlungen der SGR, Januskinase-Inhibitoren, https://www.rheuma-net.ch, Letzter Zugriff: Juni 2022.
45. Simpson EL et al. Two Phase 3 Trials of Dupilumab versus Placebo in Atopic Dermatitis. N Engl J Med, 2016. 375(24): p. 2335-2348.
46. Thaçi D et al. Efficacy and safety of dupilumab monotherapy in adults with moderate-to-severe atopic dermatitis: a pooled analysis of two phase 3 randomized trials (LIBERTY AD SOLO 1 and LIBERTY AD SOLO 2). J Dermatol Sci, 2019. 94(2): p. 266-275.
47. Blauvelt A et al. Long-term management of moderate-to-severe atopic dermatitis with dupilumab and concomitant topical corticosteroids (LIBERTY AD CHRONOS): a 1-year, randomised, double-blinded, placebo-controlled, phase 3 trial. Lancet, 2017. 389(10086): p. 2287-2303.
48. Wollenberg A et al. Tralokinumab for moderate-to-severe atopic dermatitis: results from two 52-week, randomized, double-blind, multicentre, placebo-controlled phase III trials (ECZTRA 1 and ECZTRA 2). Br J Dermatol, 2021. 184(3): p. 437-449.
49. Silverberg JI et al. Tralokinumab plus topical corticosteroids for the treatment of moderate-to-severe atopic dermatitis: results from the double-blind, randomized, multicentre, placebo-controlled phase III ECZTRA 3 trial. Br J Dermatol, 2021. 184(3): p. 450-463.
50. Bieber T. Atopic dermatitis: an expanding therapeutic pipeline for a complex disease. Nat Rev Drug Discov, 2022. 21(1): p. 21-40.