L’éventail des pneumopathies interstitielles est large. Sur le plan thérapeutique, des progrès ont été réalisés dans de nombreux domaines au cours de la dernière décennie. Dans le cas de la fibrose pulmonaire idiopathique (FPI), jusqu’au début des années 2010, il n’y avait pratiquement aucune possibilité de proposer un traitement valable aux patients concernés. Toutefois, il existe désormais des médicaments qui retardent considérablement la progression de la maladie.
En mai, la ligne directrice en langue allemande sur le diagnostic de l’ILD a été renouvelée. “La pneumopathie interstitielle est un terme que nous utilisons encore, bien que la maladie parenchymateuse diffuse des poumons serait beaucoup plus appropriée pour décrire le tableau”, a déclaré en introduction le professeur Michael Pfeifer, chef du service de pneumologie de l’hôpital universitaire de Ratisbonne (Allemagne). Il a présenté la casuistique d’un de ses patients, à partir de laquelle il a démontré comment se déroule classiquement la procédure diagnostique selon les lignes directrices. Avec une collègue, il a accordé une attention particulière au diagnostic différentiel d’un point de vue radiologique.
Le patient a été exposé à des isocyanates
Un patient de 62 ans a consulté le professeur Pfeifer en septembre 2016 pour une pneumopathie interstitielle. Au premier plan de la symptomatologie se trouvait une dyspnée d’effort, mais aussi une toux irritative plus importante. Sinon, l’homme n’a pas présenté d’anomalies significatives, ni fièvre ni perte de poids, et il n’a pas non plus présenté de troubles articulaires, de lésions cutanées ou de symptômes de siccité.
Il n’y avait pas d’exposition aux animaux domestiques ou aux moisissures dans l’environnement domestique et familial, mais le patient travaillait comme mouleur de plastique et était donc exposé aux isocyanates et aux vapeurs. Par le passé, un centre de rééducation avait déjà tenté d’obtenir la reconnaissance d’une maladie professionnelle, mais cette demande avait été refusée. L’homme avait des allergies au pollen de graminées, mais ne présentait pas d’autre profil de risque particulier. Il ne buvait pas d’alcool et ne fumait pas depuis 1989, avant cela il avait eu 10 Pack Years.

Les antécédents médicaux étaient une hypertension artérielle et de l’asthme bronchique depuis 15 ans, mais ce dernier était asymptomatique et non prédominant au moment de la présentation. Il avait un syndrome d’apnée obstructive du sommeil avec un traitement par PPCN et une opération du nez l’année précédente. Le patient prenait du vérapamil 120 mg, du valsartan 160 mg, du Symbicort 320/9 μg et du salbutamol.
L’examen a révélé un râle inspiratoire basal bilatéral, sans autre anomalie particulière que l’obésité et l’hypertension artérielle. La mesure de la fonction pulmonaire a révélé une restriction de la courbe débit-volume. TLC était de 73,5%, VC de 70,7% et FVC de 64,1%. La capacité de diffusion a montré une légère restriction.
Examen radiologique en HRCT
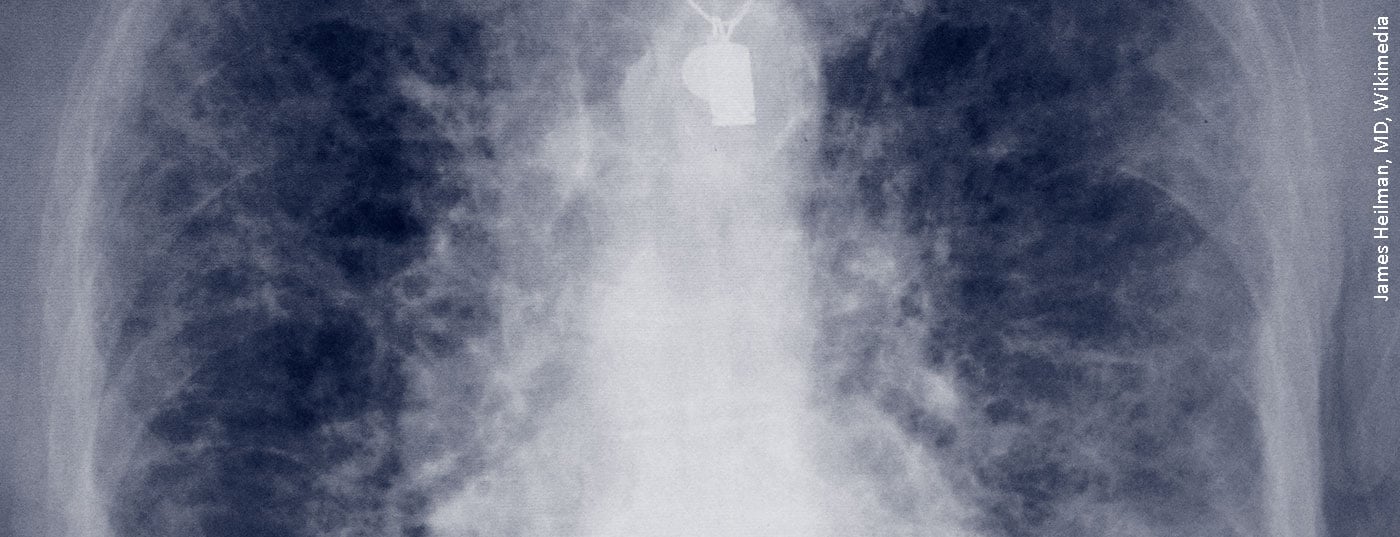
Le service de radiologie de la clinique de Donaustauf a réalisé une tomodensitométrie à haute résolution (HRCT). Le professeur Okka Hamer, chef du service de radiologie, a souligné que ce scanner en coupes fines devrait être la norme lorsqu’il s’agit d’évaluer la DLI. L’HRCT a montré des réticulations bilatérales concernant la périphérie des poumons. “Cette image serait aujourd’hui classée comme UIP probable, c’est-à-dire probable”, a déclaré le professeur Hamer, en raison des réticulations, des broncheolectasies de traction, de l’absence de rayons de miel sûrs et de l’absence de signes qui allaient à l’encontre d’un modèle UIP. Il y a quatre ans, les collègues du Board for Interstitial Pulmonopathy l’ont désigné comme possible, c’est-à-dire possible UIP (Usual Interstitial Pneumonia), selon la classification de l’époque. Mais le diagnostic différentiel avec une pneumonie interstitielle non spécifique (PINS) a également été discuté. Une cryobiopsie transbronchique et une consultation en rhumatologie ont été recommandées pour un examen plus approfondi.
Le laboratoire de rhumatologie a montré un titre ANA à 1:320, le facteur rhumatoïde était de 26,6 UI/ml (<15,0). Cependant, malgré ces anomalies, aucune indication claire de maladie rhumatismale inflammatoire systémique n’a été trouvée lors de la consultation rhumatologique.
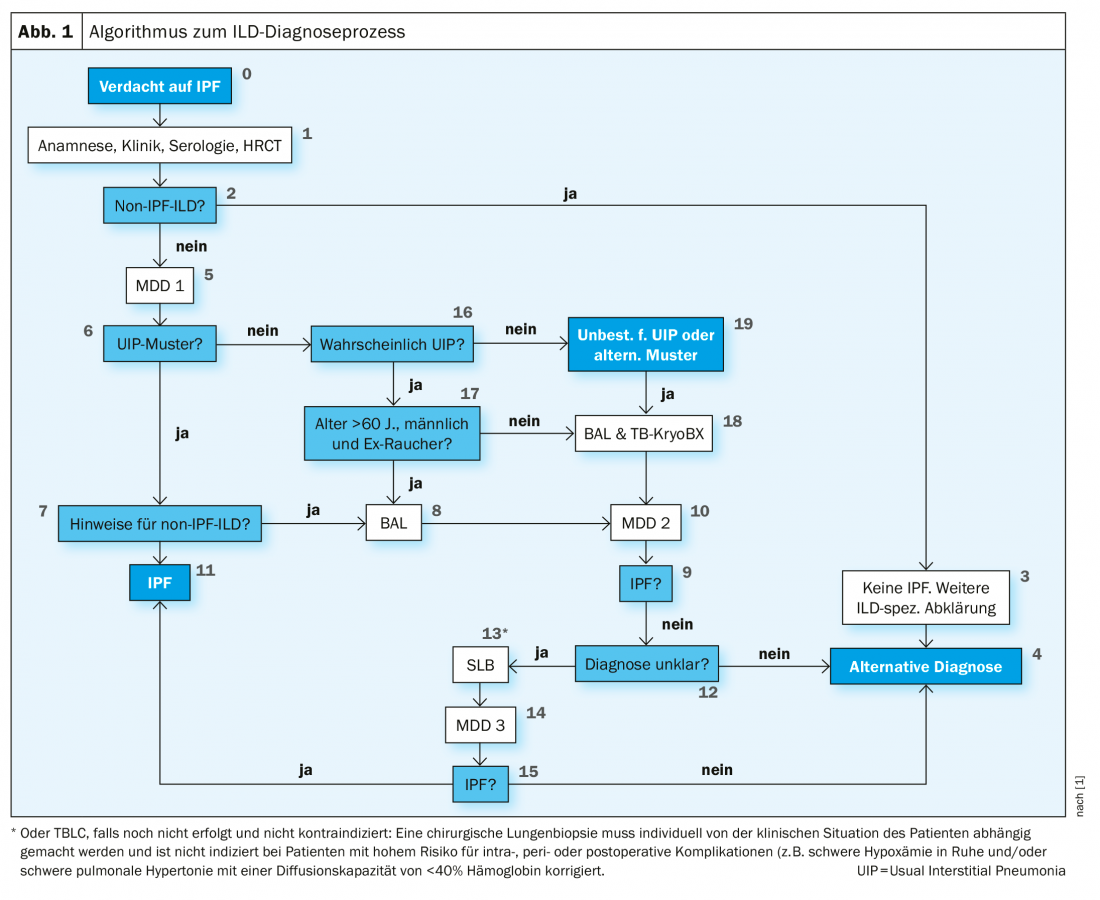
L’approche des médecins en 2016 correspond également aux directives S2k actuelles des pays germanophones (Fig. 1). Celles-ci stipulent que les patients suspectés de FPI et dont l’IRM est indéterminée ou alternative pour la PUI doivent subir un lavage broncho-alvéolaire (LBA) et une cryobiopsie. Le professeur Pfeifer a fait remarquer que, contrairement à cela, les lignes directrices internationales sont encore très critiques à l’égard de la cryobiopsie. Bien qu’elle soit mentionnée, elle n’est recommandée que dans les centres très expérimentés. Dans de nombreux pays, la VATS (chirurgie thoracoscopique vidéo-assistée) reste le moyen de premier choix.
La thérapie a apporté la stabilisation
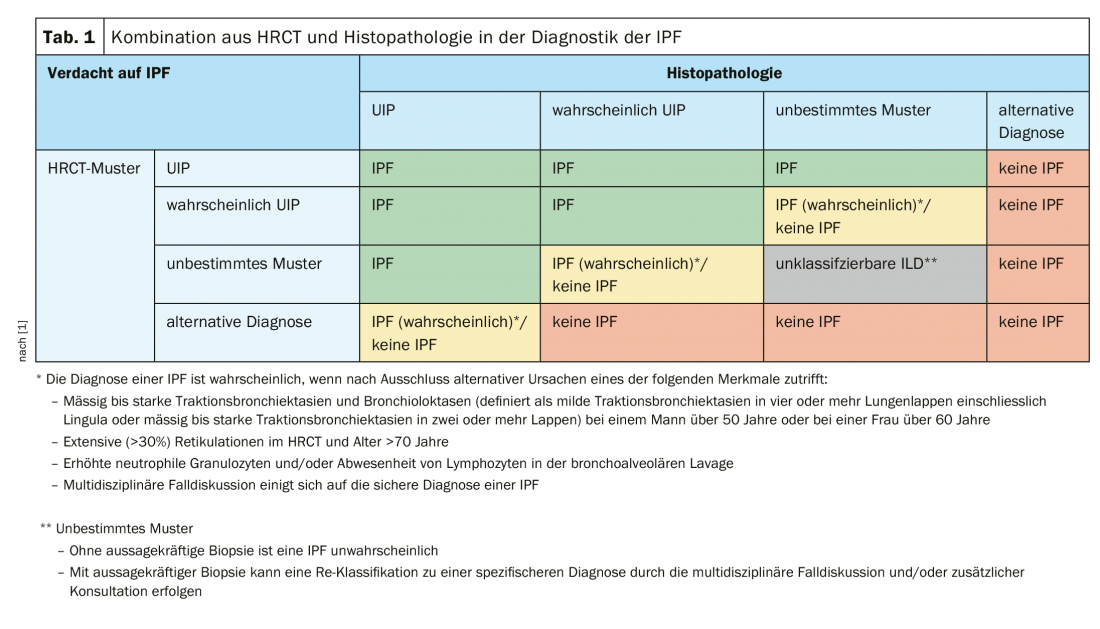
Cependant, même la cryobiopsie n’a pas permis d’obtenir une confirmation histologique chez ce patient. De même, la mise en place d’un BAL n’a pas abouti. Le conseil ILD suivant a alors décidé d’organiser un VATS. Dans ce cas, la région du lobe supérieur présentait d’une part un emphysème panazinique et d’autre part un tableau de pneumonie chronique d’hypersensibilité, peut-être dans le cadre d’une exposition professionnelle à l’isocyanate. Dans la région du lobe inférieur, l’image complète correspondait à un modèle d’UIP avec formation de nids d’abeille et de nodules de fibroblastes. Cela a finalement conduit à un troisième conseil ILD final, qui a conclu à l’existence d’une IPF. Les médecins ont recommandé l’instauration d’un traitement antifibrotique par la pirfénidone ou le nintedanib. Cette approche reflète également les recommandations des lignes directrices S2k, qui prévoient une combinaison de HRCT et d’histopathologie dans le diagnostic de la FPI (tableau 1).
Sous traitement, une nette stabilisation sans progression a été observée entre début 2017 et début 2019. Cependant, en mars 2019, le diagnostic de syndrome myélodysplasique avec excès de blastes (SMD-EB 1) a été posé chez le patient dans le cadre d’une réaction fébrile. Malgré cela, après consultation de l’hémato-oncologue, le traitement par nintedanib a été poursuivi, mais le patient est finalement décédé de la maladie. Les médecins traitants n’ont toutefois pas pu établir de lien de cause à effet entre le traitement antifibrotique et l’apparition de l’hémopathie maligne.

En résumé, il s’agit d’un cas presque classique qui permet de comprendre comment, dans le cadre du travail quotidien, il est possible de traiter un tel cas de manière structurée et conformément aux directives, afin de le classer et de mettre en place un traitement sur la base de cette classification.
Source : Séminaire en ligne “Diagnostic différentiel et traitement de la DLI”, Boehringer Ingelheim Partner’s Satellite, streamed-up.com
Littérature :
- Behr J, et al : Pneumologie 2020 ; 74(5) : 263-293 ; doi : 10.1055/a-1179-2905.
InFo PNEUMOLOGIE & ALLERGOLOGIE 2020 ; 2(3) : 28-30 (publié le 22.9.20, ahead of print)