L’amyotrophie spinale est une maladie génétique rare qui se caractérise par la dégénérescence des motoneurones de la moelle épinière et du tronc cérébral inférieur. Les experts s’accordent désormais à dire que la gestion du traitement doit être axée sur les besoins individuels et les objectifs subjectifs de la personne concernée, en particulier avec l’âge. En effet, ces derniers ont une grande influence sur la qualité de vie.
Un nouveau-né sur 10 000 environ présente un défaut génétique qui perturbe la transmission des impulsions par les motoneurones. L’amyotrophie spinale (SMA) est une maladie musculaire rare, principalement autosomique récessive, caractérisée par une dégénérescence des cellules de la corne antérieure motrice et des noyaux des nerfs crâniens moteurs. C’est la maladie héréditaire la plus fréquente entraînant la mort du nourrisson et elle est souvent diagnostiquée très tôt – mais pas exclusivement [1,2]. La SMA peut également ne se manifester qu’à l’âge adulte [3]. Cette hétérogénéité nécessite donc un traitement personnalisé qui tient compte de l’activité de la maladie et des besoins du patient.
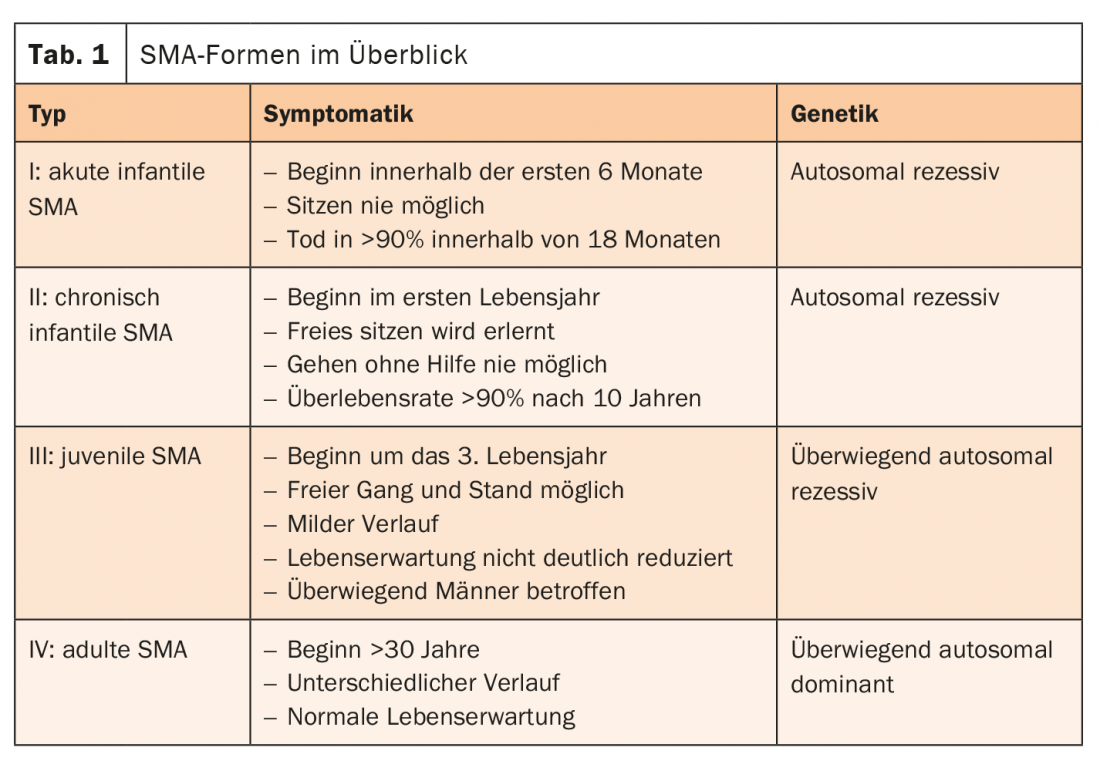
La cause de la SMA est généralement un défaut du gène SMN1. Avec le gène SMN2, il forme la protéine “Survival of Motor Neuron” (SMN). Celui-ci joue un rôle central dans la transmission des impulsions des cellules nerveuses vers les muscles. Si le gène SMN1 disparaît, la protéine importante ne peut être produite que par le gène SMN2 restant. Par conséquent, plus il y a de copies de SMN2, plus la SMA commence tard et plus l’évolution est favorable. Les patients atteints de SMA d’apparition tardive (type II-IV) ont souvent une espérance de vie normale. Les formes de SMA sont différenciées en fonction du modèle de distribution, du début de la maladie, de la sévérité et du mode d’hérédité (tableau. 1) [4].
Dépister les nouveau-nés dans la mesure du possible
Seul un examen génétique permet d’établir un diagnostic sûr de la SMA. Cependant, étant donné que la SMA de type 1, si elle n’est pas traitée, entraîne la mort ou nécessite une ventilation mécanique permanente dans 90% des cas avant l’âge de deux ans, il est indiqué de diagnostiquer rapidement la maladie et de commencer le traitement le plus tôt possible. La gestion de la thérapie est complexe et comprend, outre les soins aigus, des mesures concernant la rééducation, l’orthopédie, l’assistance respiratoire, la physiothérapie et les interventions médicamenteuses.
Succès thérapeutique également chez les adolescents et les adultes
Le premier médicament qui ne se contente pas d’agir sur les symptômes, mais s’attaque aux causes de la maladie en compensant le défaut génétique sous-jacent, a été approuvé en 2017 avec le Nusinersen. L’oligonucléotide antisens (ASO) est un modulateur d’épissage spécifique qui laisse le génome tel qu’il est et renforce la fonction naturelle de la protéine SMN2. Cela permet de produire de plus grandes quantités de protéines SMN complètes et fonctionnelles. Les premières données d’une étude de cohorte multicentrique ont maintenant été présentées. L’objectif de cette étude observationnelle non interventionnelle est avant tout d’examiner les objectifs et les attentes en matière de traitement des patients adultes atteints de 5q-SMA, ainsi que la satisfaction subjective des patients vis-à-vis du traitement [5].
Les résultats préliminaires montrent que ce sont surtout les objectifs individuels de traitement qui revêtent une importance particulière. Parmi les différents types de 5q-SMA, les objectifs du traitement varient considérablement. Ainsi, la préservation de la fonction du bras est l’un des objectifs de traitement les plus courants, avec une prédominance chez les patients de type 1 et de type 2. Chez les patients atteints de SMA de type 3, la priorité est plus souvent donnée au maintien et à l’amélioration des fonctions des jambes. Pour cela, la thérapie ASO semble appropriée. D’autres résultats d’études démontrent en outre que les adultes atteints de 5q-SMA peuvent également stabiliser différentes capacités motrices ou même bénéficier d’améliorations motrices cliniquement significatives [6-8].
Congrès : DGM 2021
Littérature :
- Bowerman M, et al : Stratégies thérapeutiques pour l’atrophie musculaire spinale : SMN et au-delà. Dis Model Mech 2017 ; 10 : 943-954.
- Borasio G, et al. : Diagnostic des atrophies musculaires spinales. Neurologie 2001 ; 20:113-118.
- www.sma-schweiz.ch/spinale-muskelatrophie/typen-der-proximalen-sma (dernier accès le 05.04.2021)
- www.muskelgesellschaft.ch/diagnosen/spinale-muskelatrophien-sma (dernier accès le 05.04.2021)
- Meyer, et al. : Congrès DGN 2020, P333.
- Hagenacker T, et al : Lancet Neurol 2020 ; 19(4) : 317-325.
- Walter MC, et al : J Neuromuscul Dis 2019 ; 6 : 453-465.
- Maggi L, et al : JNNP. 2020, Nov;91(11) : 1166-1174.
InFo NEUROLOGIE & PSYCHIATRIE 2021 ; 19(3) : 41 (publié le 5.6.21, ahead of print)
PRATIQUE DU MÉDECIN DE FAMILLE 2021 ; 16(8) : 45