Des gonflements périodiques, très douloureux et étendus des ganglions lymphatiques peuvent également être le signe d’une maladie de Castleman rare. Cette maladie rare se divise en deux catégories : la maladie de Castleman unicentrique (MCU) et les formes multicentriques. La forme multicentrique est une maladie systémique grave qui n’est pas facile à diagnostiquer.
Un gonflement des ganglions lymphatiques se produit rapidement et n’est pas inquiétant en soi. En revanche, en cas de gonflements périodiques, très douloureux et importants des ganglions lymphatiques, il faut également penser à la maladie de Castleman. Cette maladie rare est divisée en deux catégories : la maladie de Castleman unicentrique (MCD) et les formes multicentriques, et elle est souvent négligée. L’UCD est une forme bénigne de la maladie, qui se caractérise par une hyperplasie localisée du tissu lymphatique [1]. Dans ce cas, l’excision du ganglion lymphatique concerné est le traitement de choix [2]. En revanche, la situation est plus problématique dans le cas de la maladie de Castleman multicentrique (MCD). Il s’agit d’une maladie systémique grave dont le pronostic est souvent défavorable. La surproduction de différentes cytokines, en particulier l’interleukine-6 (IL-6), entraîne une symptomatologie hétérogène. Il existe une association étroite avec le virus de l’herpès humain 8 (HHV-8). Celui-ci est à son tour largement congruent avec les infections par le VIH. En conséquence, plus de 90% des patients infectés par le VIH sont également positifs pour le HHV-8. Inversement, chez les patients séronégatifs, le taux n’est que de 10 à 15% – dans ce cas, la MCD idiopathique prédomine [3] (Fig. 1).
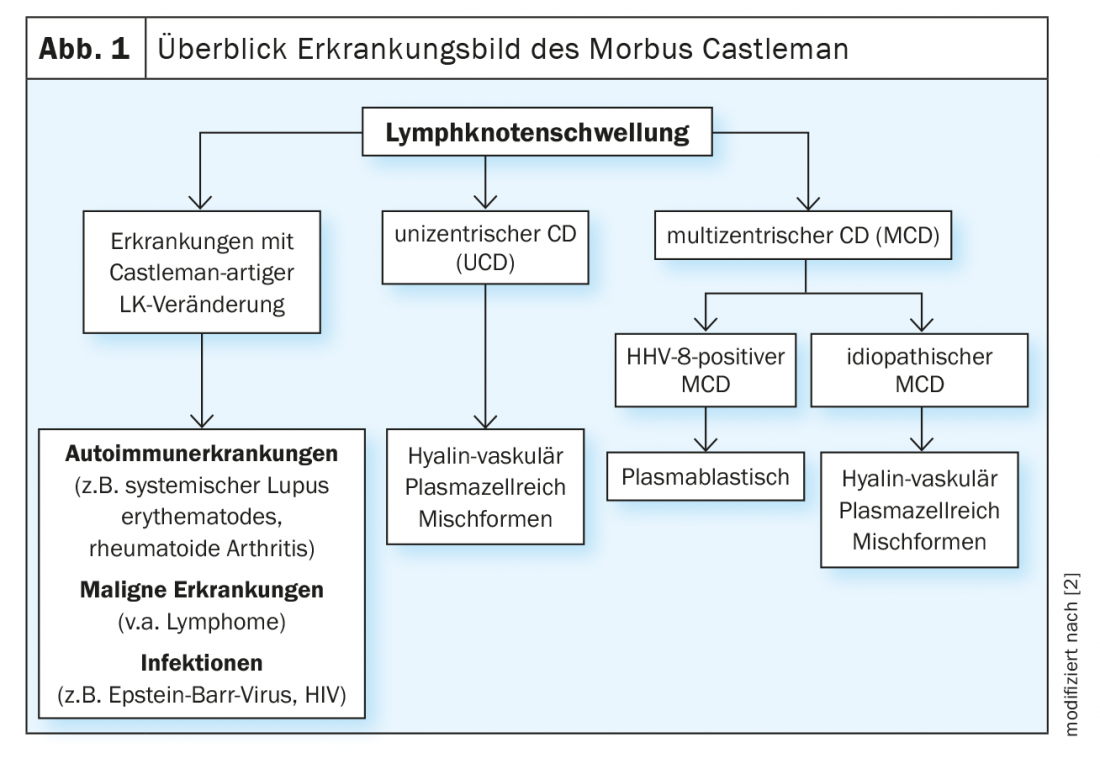
Épidémiologie
En principe, la maladie de Castleman est une maladie très rare, mais qui n’est certainement pas diagnostiquée correctement dans la plupart des cas. On estime à 2,4 le nombre de cas par million d’habitants, ce qui correspond à environ 20 cas en Suisse [4]. Il semble que le MCD (environ 35,4%) soit moins fréquent que l’UCD (64,6%) [5]. L’incidence est plus élevée chez les patients infectés par le VIH. Selon une étude britannique, elle est de 4,3 cas pour 10 000 patients-années, avec une légère tendance à la hausse ces dernières années [2,6]. Dans les études d’observation, la mortalité dans les 5 premières années après le diagnostic était de 30 à 35% [7]. Selon une revue systémique, la survie sans maladie dans la MCMI était de 45,7% à 3 ans [8].
Pathogenèse
La forme multicentrique avec augmentation de la taille de nombreux ganglions lymphatiques a été décrite pour la première fois en 1978. Elle est plus fréquente chez les patients séropositifs. Le HHV8 est responsable de la maladie de Castleman multicentrique chez presque tous les patients séropositifs et d’environ la moitié des maladies chez les patients séronégatifs [9]. Chez eux, il y a production d’une interleukine virale très similaire à l’IL-6 humaine et qui a des effets similaires. Il lui suffit de se lier à l’une des deux sous-unités du récepteur de l’IL-6 pour être actif. Cela explique l’effet beaucoup plus large sur les cellules cibles et probablement aussi les “tempêtes de cytokines” typiques [10,11,2]. L’interleukine-6 et d’autres cytokines pro-inflammatoires induisent la prolifération des cellules B et des plasmocytes, la sécrétion de facteurs de croissance endothéliaux vasculaires et l’angiogenèse.
En revanche, les causes de l’iMCD sont -actuellement- encore controversées. Les scientifiques pensent qu’une maladie inflammatoire auto-immune, une infection virale et une prédisposition génétique pourraient être responsables [12]. Outre l’IL-6, d’autres molécules de signalisation sont impliquées, notamment le VEGF (“vascular endothelial growth factor”), le TNF-alpha et l’interleukine-1 [13]. La prolifération lymphocytaire dans l’iMCD est généralement polyclonale et résulte de l’hypercytokinémie [14,2]. Cependant, une prolifération monoclonale a également été observée dans certaines lésions de sous-type vasculaire hyalin [15,2].
Symptômes et plaintes
Les symptômes se présentent de manière très hétérogène, ce qui rend souvent difficile un diagnostic efficace. En général, la maladie s’accompagne toutefois de douleurs des ganglions lymphatiques atteints. Ces gonflements surviennent souvent par poussées, en particulier dans le cas de la MCD HHV-8 positive. S’y ajoutent des symptômes B évidents tels que fièvre, sueurs nocturnes et perte de poids. Presque tous les patients se plaignent de faiblesse et d’une sensation de malaise, de nausées, de vomissements et de perte d’appétit. En outre, la rate et le foie présentent une augmentation de volume. S’y ajoutent une hépatomégalie, des symptômes respiratoires et une tendance à l’œdème en cas d’hypalbuminémie. On observe généralement une anémie sévère sur le plan hématologique, qui peut souvent s’accompagner d’une thrombocytopénie massive.
La maladie évolue typiquement par poussées qui durent de quelques jours à quelques semaines et pendant lesquelles les patients sont souvent très fiévreux et gravement malades. Entre les deux, il y a des périodes plus longues, qui peuvent durer des semaines, pendant lesquelles les patients se sentent nettement mieux. La taille des ganglions lymphatiques peut revenir pratiquement à la normale. La fréquence et l’intensité des poussées augmentent généralement au fil du temps, mais on connaît aussi des évolutions auto-limitées. Il existe cependant un risque nettement accru de lymphomes malins, en particulier de sous-types de lymphomes plutôt rares tels que les lymphomes plasmablastiques ou les lymphomes à effusion primaire [2,12].
Un iMCD se traduit par un symptôme de fatigue prononcé, surtout dans les phases précoces. Contrairement à la MCD, les symptômes tels que la fièvre, le gonflement des ganglions lymphatiques et l’anémie sont nettement moins prononcés et l’évolution par poussées est moins brutale. Les symptômes peuvent aller de légers symptômes constitutionnels à une évolution potentiellement mortelle avec anasarque et défaillance de plusieurs organes. Un sous-type d’iMCD gravement évolutif, le syndrome TAFRO, a été redéfini. Il s’agit d’un ensemble de symptômes comprenant une thrombocytopénie, une ascite, de la fièvre, une fibrose réticulée de la moelle osseuse et une organomégalie avec une γ-globuline normale [16]. En outre, des associations avec le syndrome POEMS, un tableau clinique associant une neuropathie périphérique, une organomégalie, une endocrinopathie, une gammapathie monoclonale et des lésions cutanées, sont fréquemment observées [2].
Diagnostic
La maladie de Castleman doit faire l’objet d’un diagnostic différentiel avec les lymphomes et autres maladies graves. La forme sévère, en particulier, est souvent prise pour un lymphome. L’examen histopathologique d’un ganglion lymphatique extirpé est donc obligatoire. Une ponction ne suffit généralement pas. La préparation montre une lymphadénite réactive avec prolifération de plasmocytes dans la pulpe et des modifications régressives des centres germinaux, qui sont généralement stratifiés en pelures d’oignon et traversés par des vaisseaux. Dans le cas d’un MCD HHV-8 positif, on observe généralement une image plasmablastique, qui présente souvent un motif dit de “mite” au niveau de la zone du manteau. Le motif caractéristique en pelure d’oignon est souvent partiellement détruit. La détection du HHV-8 se fait par coloration pour le LANA-1 (“latency-associated-nuclear-antigen”). Le sang prélevé pendant une poussée de la maladie montre des taux élevés d’IL-6 et de CRP. La triade symptômes B, virémie HHV-8 et résultats histologiques permet d’orienter le diagnostic [17].
Le diagnostic de l’iMCD est beaucoup plus difficile, car il faut exclure un grand nombre d’autres maladies. Les modifications histopathologiques se retrouvent également, par exemple, dans un grand nombre d’infections (dont le virus Epstein-Barr) ou encore dans le lupus érythémateux, la polyarthrite rhumatoïde, le syndrome de Sjögren et certaines néoplasies, dont principalement les lymphomes malins. Outre les modifications histopathologiques, il faut donc toujours prendre en compte les paramètres cliniques et de laboratoire. On distingue trois sous-types histopathologiques d’iMCD : le type hyalin-vasculaire, le type plasmocytaire et le type mixte. Tous se produisent dans environ 20 à 40% des cas [18]. Le type hyalin vasculaire se caractérise par des cellules folliculaires dendritiques (FDC) dysplasiques et des centres germinaux atrophiques traversés par des vaisseaux hyalinisés, autour desquels s’organisent des lymphocytes concentriques. Dans le type plasmocytaire, les centres germinaux sont plus hyperplasiques qu’atrophiques et les FDC ainsi que l’architecture des ganglions lymphatiques sont normaux [2].
Thérapie
La maladie de Castleman unicentrique a un très bon pronostic, car une intervention chirurgicale permet de retirer le ganglion lymphatique atteint. La récidive de la maladie est extrêmement rare. Cependant, comme la MCD touche de nombreux ganglions lymphatiques, l’intervention chirurgicale n’est pas efficace. Le traitement reste difficile. Il n’existe pas de traitement standard pour les patients atteints de la maladie de Castleman multicentrique. Conceptuellement, des substances immunosuppressives et cytotoxiques sont utilisées. Les glucocorticoïdes sont efficaces, mais n’entraînent souvent pas d’amélioration des symptômes à long terme (figure 2).
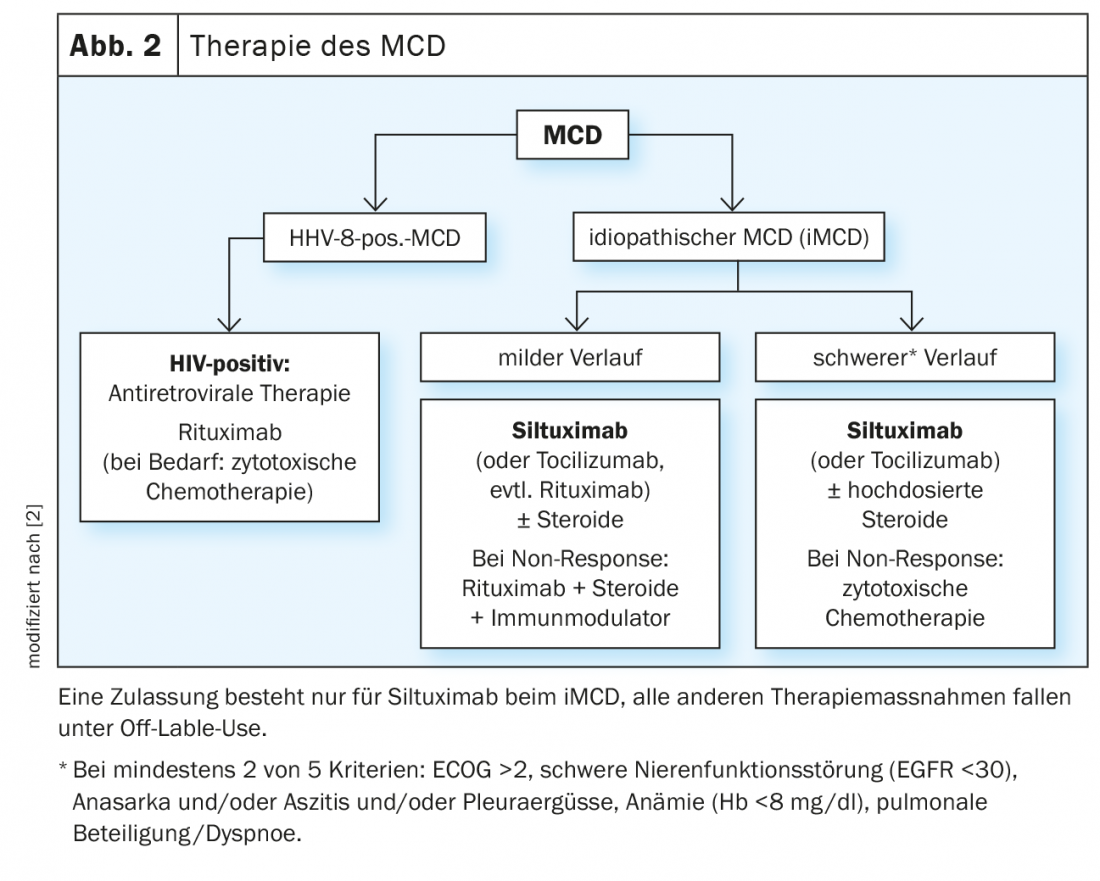
Dans le cas de l’iMCD, on utilise principalement le blocage de la voie de signalisation de l’IL-6 au moyen d’anticorps monoclonaux comme le siltuximab. Dans le cas de la MCD HHV-8 positive, on mise sur l’élimination cytotoxique des cellules responsables de la surproduction de cytokines. Les traitements immunomodulateurs sont principalement des stéroïdes, ainsi que des substances antivirales comme le valganciclovir. Depuis 2018, il existe pour la première fois des recommandations basées sur un consensus pour l’iMCD [19]. Elles sont basées sur l’expérience de 344 patients ayant suivi 479 thérapies au total, tiennent compte de la gravité de la maladie, des thérapies antérieures et de la réponse.
Le siltuximab, un anticorps monoclonal dirigé contre l’IL-6 humaine, est le seul médicament autorisé à ce jour pour le traitement de la MICI. Il est utilisé dans toutes les formes de MICI, avec ou sans stéroïdes. Dans l’étude pivot, 79 patients atteints d’iMCD ont été traités pendant trois semaines soit par siltuximab, soit par placebo [20,7]. Le critère d’évaluation principal était une réponse durable en termes de taille de la tumeur et d’amélioration d’un score de symptômes cliniques pendant au moins 18 semaines. Alors qu’aucune réponse n’a été observée dans le groupe placebo, les taux étaient de 34% dans le groupe placebo. Le siltuximab a été globalement bien toléré. Les effets secondaires fréquents et caractéristiques sont le prurit cutané, les infections des voies respiratoires supérieures, l’exanthème et l’œdème local, principalement de sévérité 1 et 2. Les effets secondaires graves sont la fatigue (12%) et les sueurs nocturnes (8%). Cependant, ce taux n’était pas plus élevé que celui observé avec le placebo, de sorte que ces symptômes sont plutôt à attribuer à la maladie sous-jacente [7]. Des données d’études récentes montrent désormais que les patients atteints de MCD nouvellement diagnostiqués et ceux qui ont déjà été traités répondent de la même manière à la substance [21]. Au total, 46 patients prétraités et 33 patients nouvellement diagnostiqués ont été randomisés pour recevoir soit l’anticorps (n=53), soit le placebo (n=26). A l’exception du sous-type histologique, il n’y avait pas de différences significatives dans les caractéristiques de la ligne de base. La durée médiane de traitement a été de 375 et de 375 jours respectivement. 233 jours. Les taux de réponse tumorale durable et de réponse symptomatique chez les patients traités par siltuximab, comparés au placebo, étaient de 34,5% vs 0% chez les patients précédemment traités et de 33,3% vs 0% chez les patients nouvellement diagnostiqués. Le temps médian jusqu’à l’échec du traitement (TTF) n’a pas été atteint avec le siltuximab dans les deux sous-groupes.
Le tocilizumab, un anticorps dirigé contre le récepteur IL-6, est considéré comme une alternative au siltuximab dans les recommandations internationales, mais n’est autorisé en Europe que dans la polyarthrite rhumatoïde.
Le rituximab, un anticorps monoclonal anti-CD20, n’est pas non plus autorisé pour le traitement de la MCD. Les patients atteints de MCD HHV-8 positive, en particulier, semblent bien y répondre. Dans plusieurs séries de cas, une amélioration significative de la survie globalea et de la survie sans maladie a été observée. Les cytokines, mais aussi la CRP, les immunoglobulines et la charge virale HHV-8 ont diminué [22,2]. Cependant, dans un peu plus d’un tiers des cas, il y a eu une progression du sarcome de Kaposi, qui est souvent associé à la maladie. Dans ce cas, une combinaison avec des substances cytostatiques actives sur le KS pourrait être utile [23]. Dans les situations où le pronostic vital est engagé, une splénectomie doit être envisagée. Elle réduit le pool de cellules infectées par le HHV-8 [2].
Messages Take-Home
- La maladie de Castleman est divisée en forme bénigne unicentrique (UCD) et en maladie de Castleman multicentrique (MCD).
- La MCD est une maladie systémique qui n’est souvent pas reconnue en raison de son hétérogénéité et de sa rareté.
- Les poussées s’accompagnent d’un gonflement douloureux des ganglions lymphatiques, souvent accompagné de symptômes B, tels que fièvre ou sueurs nocturnes.
- Le siltuximab est actuellement le seul médicament autorisé pour le traitement de la MCD idiopathique.
Littérature :
- Castleman B, Towne VW : Case records of the Massachusetts General Hospital ; exercices hebdomadaires de clinicopathologie. N Engl J Med 1954 ; 251 : 396-400
- Hoffmann C, Tiemann M : Maladie de Castleman multicentrique : rarement correctement diagnostiquée. Dtsch Arztebl 2019 ; 116(46) : [32]; DOI : 10.3238/PersOnko.2019.11.15.06
- Hoffmann C : La maladie de Castleman multicentrique (MCD) – une pathologie rare et souvent méconnue. Trillium 2015. www.trillium.de/zeitschriften/trillium-krebsmedizin/archiv/ausgaben-2015/heft-12015/multizentrischer-morbus-castleman-mcd-ein-seltenes-oft-verkanntes-krankheitsbild.html (dernier accès le 06.04.2020)
- Robinson D Jr, Reynolds M, Casper C, et al : Clinical epidemiology and treatment patterns of patients with multicentric Castleman disease : results from two US treatment centres. Br J Haematol 2014 ; 165 : 39-48.
- Haap M, Wiefels J, Horger M, et al : Clinical, laboratory and imaging findings in Castleman’s disease – The subtype decides. Blood Rev 2018 ; 32 : 225-234.
- Powles T, Stebbing J, Bazeos A, et al : Le rôle de la suppression immunitaire et du HHV-8 dans l’augmentation de l’incidence de la maladie de Castleman multicentrique associée au VIH. Ann Oncol 2009 ; 20 : 775-779.
- www.dgho.de/publikationen/stellungnahmen/fruehe-nutzenbewertung/siltuximab/siltuximab-dgho-stellungnahme-20141006.pdf (dernier accès le 06.04.2020)
- Talat N, Schulte KM : Maladie de Castleman : analyse systématique de 416 patients de la littérature. Oncologist 2011 ; 16:1316-24
- Fajgenbaum DC, van Rhee F, Nabel CS : HHV-8-negative, idiopathic multicentric Castleman disease : novel insights into biology, pathogenesis and therapy. Blood 2014 ; 123 : 2924-2933. DOI: 10.1182/blood-2013-12-545087.
- Li H, Wang H, Nicholas J. Détection de la liaison directe de l’interleukine-6 codée par l’herpèsvirus humain 8 (vIL-6) aux deux récepteurs gp130 et IL-6 (IL-6R) et identification des résidus d’acides aminés de vIL-6 importants pour la signalisation dépendante et indépendante de l’IL-6R. J Virol 2001 ; 75 : 3325-3334.
- Moore PS, Boshoff C, Weiss RA, Chang Y : Mimétisme moléculaire des gènes de cytokines et de voies de réponse aux cytokines humaines par KSHV. Science 1996 ; 274 : 1739-1744.
- https://medlexi.de/Morbus_Castleman (dernier accès le 06.04.2020)
- Pierson S, Stonestrom A, Ruth J, et al : Quantification of plasma proteins from idiopathic multicentric Castleman disease flares and remissions reveals ‘chemokine storm’ and separates clinical subtypes (abstract). Blood 2017 ; 130(Suppl. 1).Abstract 3592
- Ohyashiki JH, Ohyashiki K, Kawakubo K, et al. Analyses moléculaires génétiques, cytogénétiques et immunophénotypiques de la maladie de Castleman du type plasmocytaire. Am J Clin Pathol 1994 ; 101 : 290-295.
- Chang KC, Wang XC, Hung YL : Monoclonalité et anomalies cytogénétiques dans la maladie de Castleman vasculaire hyaline. Mod Path 2014 ; 7 : 823-831.
- Iwaki N, Fagenbaum D, Nabel CS, et al : Clinicopathologic analysis of TAFRO syndrome demonstrates a distinct subtype of HHV-8-negative multicentric Castleman disease. Am J Hematol 2016 ; 91 : 220-226.
- Bower M, Pria AD, Coyle C, et al : Diagnostic criteria schemes for multicentric Castleman disease in 75 cases. J Acquir Immune Deficit Syndr 2014 ; 65(2) : e80-82.
- Liu AY, Nabel CS, Finkelman BS, et al : Idiopathic multicentric Castleman’s disease : a systematic literature review. Lancet Hematol 2016 ; 3 : e163-175.
- van Rhee F, Voorhees P, Dispenzieri A, et al : International, evidence-based consensus treatment guidelines for idiopathic multicentric Castleman disease. Blood 2018 ; 132 : 2115-2124.
- van Rhee F, Wong RS, Munshi N, et al : Siltuximab for multicentric Castleman’s disease : a randomised, double-blind, placebo-controlled trial. Lancet Oncol 2014 ; 15 : 966-974, DOI : 10.1016/S1470-2045(14)70319-5.
- van Rhee F, Rossi J, Simpson D et al. Les patients atteints de la maladie de Castleman multicentrique récemment diagnostiqués ou précédemment traités répondent de manière équivalente au siltuximab. Br J Haematol. 2021 ; 192(1):e28-e31.
- Bower M, Veraitch O, Szydlo R, et al : Cytokines changes during rituximab therapy in HIV-associated multicentric Castleman disease. Blood 2009 ; 113 : 4521-4524.
- Uldrick TS, Polizzotto MN, Aleman K, et al : Rituximab plus doxorubicine liposomale chez les patients infectés par le VIH atteints de la maladie de Castleman multicentrique associée au KSHV. Blood 2014 ; 124 : 3544-3552.
InFo ONKOLOGIE & HÉMATOLOGIE 2022 ; 10(2) : 10-13