
Comment les inhibiteurs de kinase peuvent-ils être utilisés à bon escient ? Quels sont les avantages et les inconvénients d’une combinaison ? Comment le séquençage génomique peut-il contribuer à une approche thérapeutique individualisée avec de tels inhibiteurs et quels modèles d’études devraient être utilisés pour le vérifier ? Quelles leçons peut-on tirer des échecs des études et des processus de développement des inhibiteurs de kinase connus ? Toutes ces questions ont été discutées lors du congrès ESMO à Amsterdam.
Annette K. Larsen, Paris, a posé la question de savoir s’il fallait combiner les inhibiteurs de kinase et comment :
“Le développement de nouveaux agents anticancéreux qui ciblent les voies de signalisation oncogènes représente une avancée conceptuelle majeure. Cependant, les résultats cliniques sont souvent restés en deçà des attentes, en partie à cause de mutations en aval, de boucles de rétroaction inattendues ou de ce que l’on appelle le cross-talk des voies de signalisation”.
C’est pourquoi on se concentre aujourd’hui sur le ciblage de plusieurs chemins de signaux en même temps ou sur différentes étapes du même chemin de signal. Souvent, les inhibiteurs de kinase ont été ajoutés directement à des agents cytotoxiques établis sans ajustement de la dose, ce qui a parfois entraîné des effets secondaires toxiques importants. Dans son exposé, Larsen a présenté les possibilités et les limites des combinaisons d’inhibiteurs de kinases.
Deux études récentes de phase III (PACCE et CAIRO2) ont évalué l’ajout d’anticorps monoclonaux ciblant l’EGFR (mAbs), le cetuximab et le panitumumab respectivement, au bevacizumab plus chimiothérapie chez des patients atteints de cancer colorectal (CRC). Cette combinaison était non seulement associée à une survie sans progression plus courte, mais aussi à une moins bonne qualité de vie, également chez les patients atteints d’une tumeur CRAS de type sauvage.
“Pourquoi la combinaison des deux AcM n’a-t-elle pas fonctionné ? D’une part, la combinaison des agents cibles était trop toxique, d’autre part, elle n’est pas active, c’est-à-dire que les deux inhibent les ligands ou les récepteurs extracellulaires, mais ont des effets limités, voire aucun effet, sur la signalisation de la tyrosine kinase (RTK) du récepteur”, explique Larsen. Elle conclut que si l’association d’agents ciblant le VEGF et l’EGFR est possible, elle ne doit pas nécessairement être utilisée en même temps que la chimiothérapie. Le statut mutationnel de KRAS joue ici un rôle surtout pour les AcM ciblés sur l’EGFR.
Approches thérapeutiques individualisées
Emile E. Voest, Utrecht, a parlé des possibilités d’intégrer le séquençage génomique dans la décision thérapeutique : “Des informations détaillées sur la façon dont une tumeur se développe génétiquement permettent de mieux cibler certains patients pour un traitement donné. Des exemples tels que le trastuzumab pour l’expression de HER2 dans le cancer du sein, l’imatinib pour les translocations de BCR-ABL dans la leucémie, le vémurafénib et le crizotinib pour les mutations de V600E dans le mélanome et les translocations de ALK-EML4 dans le cancer du poumon ont clairement démontré la validité de l’application de ce concept. Ces succès spectaculaires sont ternis par le fait qu’ils sont temporaires, car les résistances se développent souvent”.
Grâce aux progrès de la technologie des séquences, il est désormais possible de générer des informations détaillées sur les anomalies génomiques d’une tumeur. Les analyses de gènes uniques (i.e. BRAF, KRAS) seront remplacées dans un avenir proche par des analyses de génomes entiers. Cette approche globale permet d’obtenir des informations au niveau des voies de signalisation plutôt qu’au niveau des gènes individuels. “Par exemple, chez les patientes atteintes d’un cancer du sein, nous n’avons pas trouvé de corrélation entre le succès d’une chimiothérapie et les mutations spécifiquement traçables dans le gène PI3K, mais nous en avons trouvé une avec les mutations dans la voie de signalisation PI3K”, explique le professeur Voest.
L’hétérogénéité tumorale est une dimension importante de la croissance tumorale et une excroissance clonale de populations résistantes se produit souvent pendant le traitement. Dans ce cas, les méthodes de “séquençage (ultra) profond” peuvent aider à détecter et à identifier les clones à un stade précoce, ce qui permet une approche thérapeutique anticipative. Actuellement, on pense qu’il sera plus facile de trouver un profil génétique prédictif pour les médicaments ayant un mécanisme d’action spécifique (comme le vémurafénib) que pour les inhibiteurs de kinase à large spectre (comme le sunitinib). Si l’on tient compte des conclusions sur la corrélation mentionnée ci-dessus entre le succès de la chimiothérapie et la voie PI3K, cette approche risque d’être démasquée comme étant un malentendu.
“Nous sommes à l’aube d’une ère dans laquelle des tests génétiques complets de la tumeur et de l’ADN germinal feront partie du diagnostic régulier des patients atteints de cancer. L’interprétation compétente des données extrêmement complexes sera notamment décisive pour le choix correct d’un traitement basé sur l’ADN. Pour cela, il faut de grandes bases de données qui offrent la possibilité de relier le succès clinique aux données génétiques”, a conclu le professeur Voest.
Études de génotype vs. études de panier
“Les études génotypiques sont basées sur l’analyse du génotype d’un certain nombre de patients atteints d’une maladie, puis sur l’attribution de différents médicaments ciblés en fonction de leur profil”, explique José Baselga, MD, New York (Fig. 1). “Les problèmes de cette approche sont les suivants : Les médicaments utilisés dans chaque groupe ne sont généralement pas les meilleurs de leur classe, mais ceux qui sont disponibles (par exemple, la première étude BATTLE a utilisé le sorafenib comme inhibiteur de RAF). De plus, si la randomisation est utilisée, il pourrait être contraire à l’éthique de randomiser certains patients au cours de l’étude, qui ne recevraient alors pas le traitement correspondant à leur profil. De plus, le nombre total de participants étant généralement faible, ce type de conception ne permet souvent pas d’acquérir suffisamment de patients présentant des mutations rares pour comparer valablement le succès clinique au profil génétique (par exemple, BRAF dans le cancer du poumon ou mutations ERBB2 dans le cancer de l’ovaire)”.
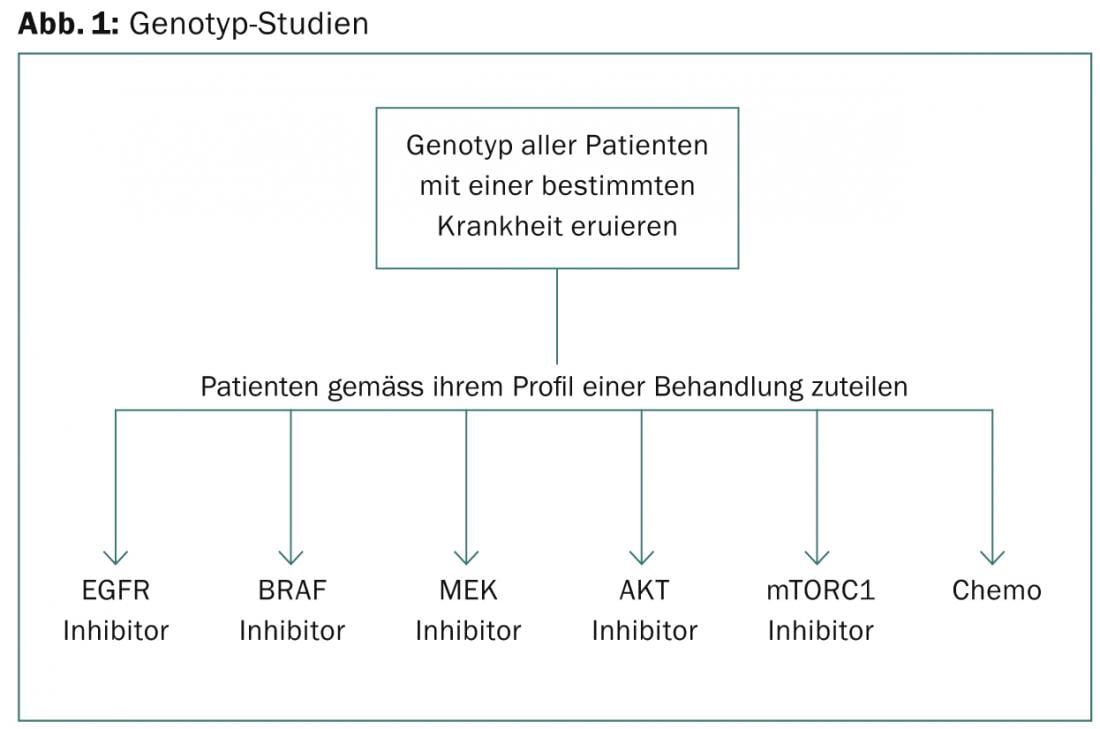
Les soi-disant. Les études de paniers permettent au contraire de tester une hypothèse concrète, par exemple : Les patients atteints de cancer de la vésicule biliaire dont la tumeur présente des mutations BRAF(V600E) répondent-ils au vémurafénib ? De plus, comme ce type d’étude est basé sur une collection de cohortes spécifiques à une maladie, il permet d’étudier l’influence de l’ascendance sur la réponse aux médicaments. De plus, les prélèvements de tissus peuvent expliquer l’hétérogénéité de la réponse. La principale critique adressée aux études par paniers est qu’elles risquent de passer à côté des patients qui pourraient répondre, mais qui ne présentent pas le biomarqueur recherché. En outre, l’identification des participants reste un obstacle : selon Baselga, il est donc impératif de séparer le protocole de dépistage du protocole de traitement.
Apprendre de ses erreurs
Comment pouvons-nous tirer des enseignements des échecs des études sur les inhibiteurs de kinase ? C’est la question que s’est posée le Dr Stefan Sleijfer, Rotterdam : “Ces dernières années, plusieurs nouveaux inhibiteurs de kinase ont été rapidement approuvés avec succès grâce aux résultats parfois sensationnels de leurs études. Mais à l’inverse, de nombreuses molécules de ce type ont également échoué, parfois dès le stade clinique précoce, parfois seulement après la réalisation d’études de phase III étendues et coûteuses. Les deux processus de développement, ceux qui ont réussi et ceux qui ont échoué, permettent de tirer de précieuses conclusions pour la conception des études futures”.
Les inhibiteurs de kinase les plus efficaces sont ceux qui ciblent et inhibent directement le produit d’un gène muté, c’est-à-dire qui ciblent un sous-groupe spécifique d’une maladie. Une fois le mécanisme d’action de ces médicaments connu, et donc les patients éligibles ciblés, les molécules ont été testées cliniquement dès le début dans des groupes de patients spécifiquement sélectionnés. Des exemples de succès sont l’imatinib dans les tumeurs stromales gastro-intestinales malignes (GIST), le vémurafénib dans les mélanomes mutés BRAF(V600E), ou le crizotinib dans le cancer du poumon non à petites cellules (NSCLC) avec gène de fusion EML4-ALK.
Une caractéristique commune importante des médicaments qui ont échoué ou qui n’ont obtenu leur autorisation de mise sur le marché qu’après un parcours difficile et coûteux est que leur mécanisme d’action était inconnu avant le début des essais cliniques. Par conséquent, aucun profil prédictif n’a pu être établi, les patients susceptibles d’y répondre n’ont pas pu être identifiés a priori et les essais cliniques ont été menés sur des populations de patients non sélectionnées. Les exemples actuels sont les antagonistes de l’IGF-1R et les inhibiteurs de mTOR.
“La principale leçon à tirer de ces études est donc l’utilité et la nécessité de découvrir le mécanisme d’action exact du médicament à l’étude dans des cadres précliniques et d’identifier des marqueurs prédictifs avant de lancer de grands essais cliniques. Si le mécanisme n’est pas connu, on peut se demander si le médicament en question doit entrer dans le processus de développement. En particulier parce que cela nécessite des ressources et des fonds extrêmement importants qui pourraient être mieux utilisés pour d’autres molécules à tester. Si un médicament dont le mécanisme d’action est inconnu est déjà en cours de développement, il est possible d’utiliser des designs adaptatifs. Par exemple, la collecte de biomatériaux est essentielle pour découvrir rétrospectivement des profils prédictifs, comme cela a été le cas pour les inhibiteurs de kinase EGFR dans le NSCLC”, a résumé le Dr Sleijfer.
Source : “Optimal use of Targeted Kinase Inhibitors (TKIs)”, symposium au congrès ESMO, 27 septembre – 1er octobre 2013, Amsterdam
InFo ONKOLOGIE & HÄMATOLOGIE 2014 ; 2(1) : 45-47









