Le sindromi da antisintetasi sono un sottogruppo delle miopatie infiammatorie idiopatiche e rappresentano circa il 35-40% dei casi. Tuttavia, se individuata in tempo, la prognosi a lungo termine per le persone colpite è relativamente buona. Un esperto ha spiegato a quali sintomi prestare attenzione e quali sono le opzioni terapeutiche.
Le sindromi da antisintetasi (ASD) sono malattie rare con una prevalenza di 1,5-9/100.000 e un’incidenza di 1,25 -2,5/1 milione. L’età media di insorgenza è compresa tra 48 e 55 anni, con un ampio intervallo alla prima manifestazione (da 20 a 80 anni). Le donne sono colpite più spesso degli uomini.
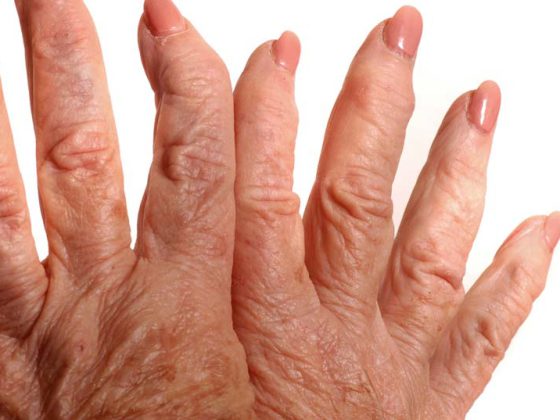
La malattia è definita dalla presenza sierologica di un anticorpo antisintetasi e clinicamente dalla combinazione di almeno uno dei tre sintomi principali: miosite, ILD o artrite, ha spiegato la dottoressa Jutta Bauhammer, Baden-Baden (D). Inoltre, possono manifestarsi sintomi secondari come febbre, sindrome di Raynaud o segni cutanei come le cosiddette mani del meccanico (Fig. 1) . All’inizio della malattia, spesso solo alcuni di questi sintomi organici sono presenti contemporaneamente. Una triade completa di miosite, coinvolgimento polmonare e artrite è più comune nella sindrome Jo-1, dove è presente solo nel 20% delle prime manifestazioni. Nel decorso successivo, circa il 50% è poi colpito, il follow-up è massimo. 16 anni.

Diagnostica di laboratorio
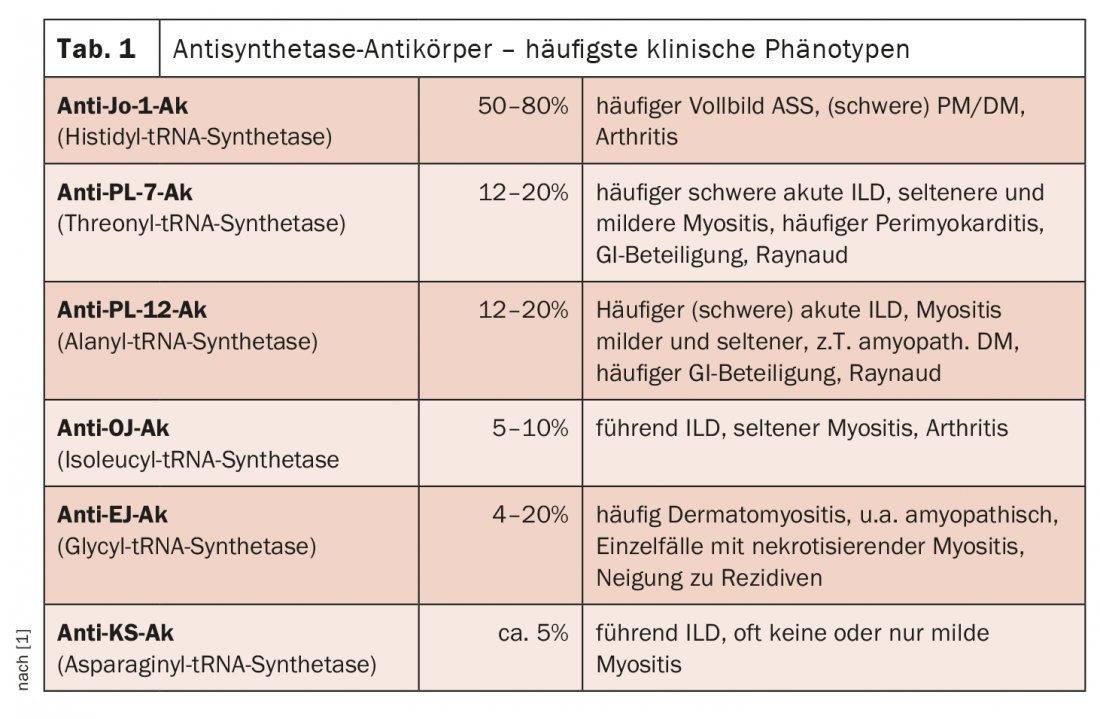
Dal punto di vista chimico-laboratoristico, sono noti 11 anticorpi antisintetasi (ASS-Ak), ma finora solo 8 di questi sono stati assegnati al quadro clinico di una sindrome antisintetasi (Tab. 1). Gli antigeni target sono le aminoacil-tRNA sintetasi e, a seconda di quale rappresenta esattamente l’antigene, esistono diversi anticorpi. Queste sintetasi sono enzimi presenti nel citoplasma e coinvolti nella traduzione; catalizzano il legame di un amminoacido al suo tRNA specifico. In genere, c’è una sola ASS-Ak per paziente, e sono mutuamente esclusive. La presenza simultanea di più ASS-Ak è considerata una rarità. L’ASS-Ak più comune nei pazienti caucasici è l’anticorpo anti-Jo-1, che è presente nel 50-80% degli individui affetti ed è diretto contro l’istidil-tRNA sintetasi. In questo caso, è più comune vedere una sindrome da antisintetasi in piena regola (cioè sono presenti tutti e tre i sintomi principali, con coinvolgimento muscolare, polmonare e articolare), con una miosite spesso grave e, in molti casi, un coinvolgimento articolare più grave con artrite manifesta.
Partecipazioni in organi
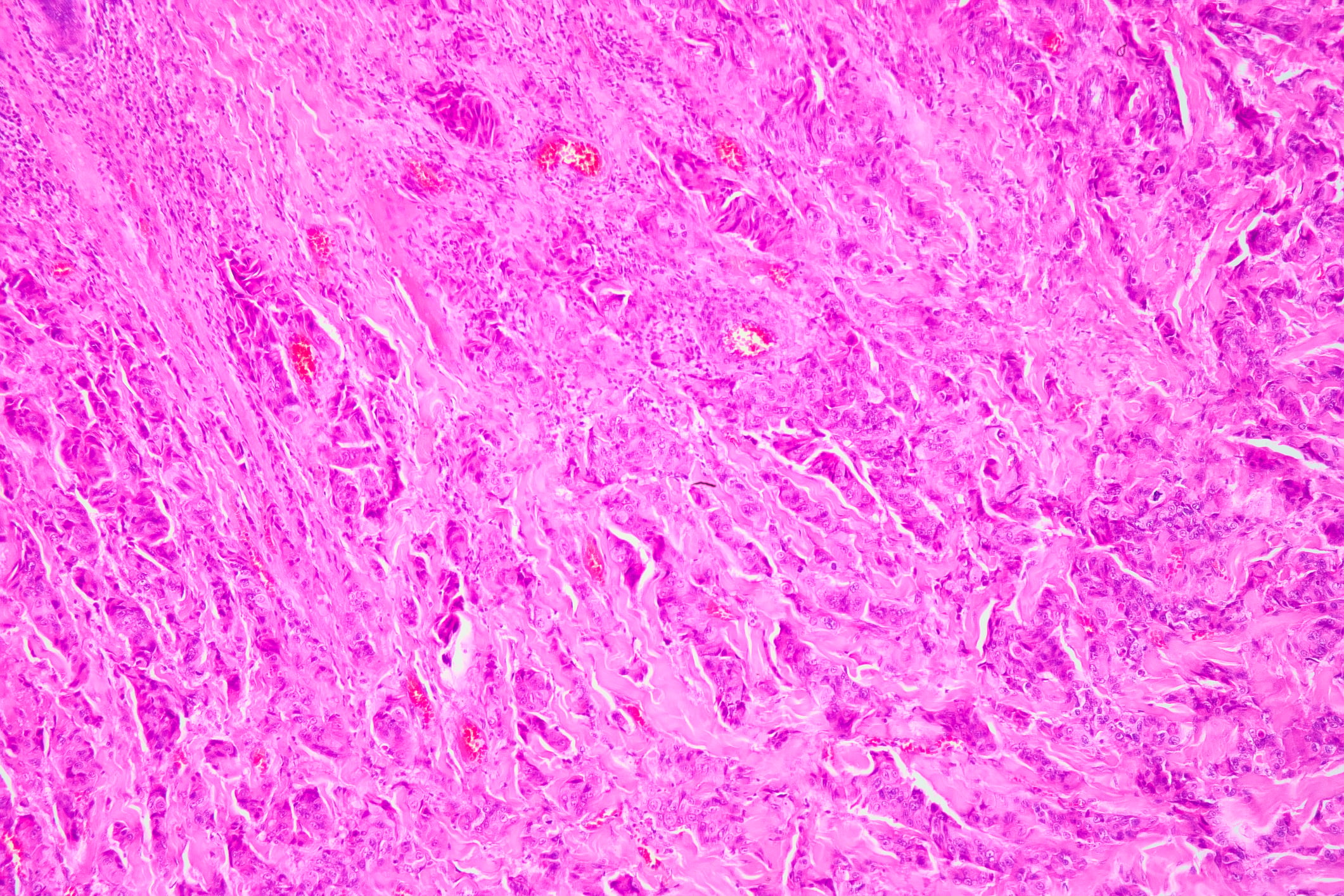
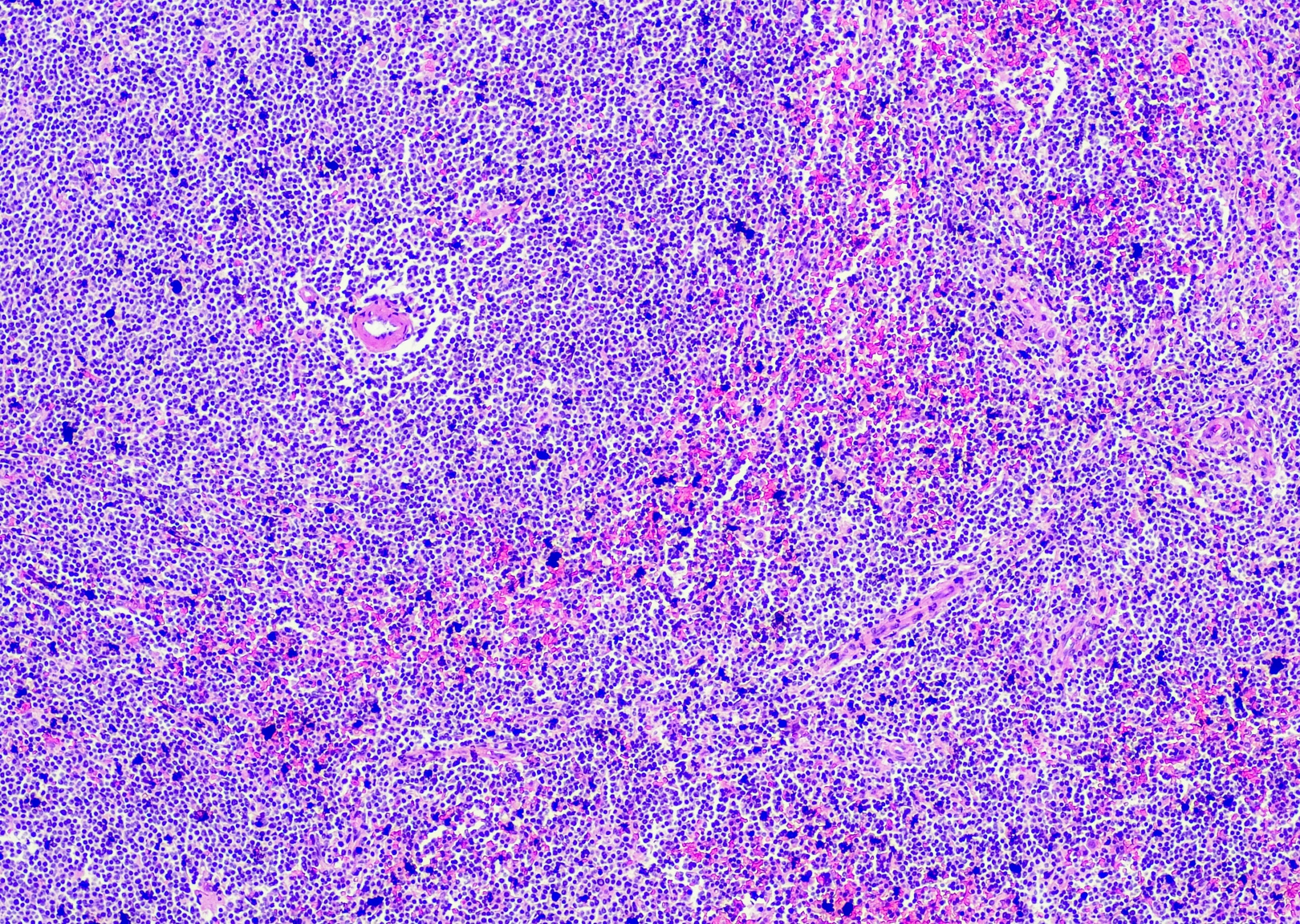
La miosite è presente nel 75-80% dei casi di Jo-1-Ak e in circa il 60% dei casi di altri anticorpi (dove il polmone è spesso l’organo principale). Clinicamente, il quadro è spesso quello della polimiosite, più raramente della dermatomiosite, e la miocardite concomitante è presente nel 3-4% dei pazienti. L’istologia mostra tipicamente un’infiammazione dominante del tessuto connettivo perimisiale con un infiltrato di macrofagi abbondanti CD68-positivi come marcatore (“miosite dominata da macrofagi”); può verificarsi una necrosi perifascicolare, mentre la densità capillare è solitamente irrilevante. Esistono anche rapporti di casi singoli di miosite necrotizzante.
Il coinvolgimento delle articolazioni si verifica nel 70% dei Jo-1-Ak e in circa il 50% dei non-Jo-1-Ak. Per gli anticorpi Jo-1, rappresenta la prima manifestazione isolata dell’organo nel 20-35%. Nella maggior parte dei casi è poliarticolare-simmetrica (70%), con coinvolgimento anche delle articolazioni piccole e medie, più raramente oligoarticolare (30%). Alcuni pazienti presentano anche artralgie infiammatorie primarie. A differenza dell’artrite reumatoide (RA), questi pazienti presentano la sindrome di Raynaud molto più frequentemente (30-50%, i pazienti RA circa il 10%). “Ora la cosa si fa ancora più insidiosa”, ha avvertito il dottor Bauhammer: in rari casi, i pazienti con sindrome antisintetasica possono avere anche anticorpi anti-CCP positivi, che in realtà sono classificati come molto specifici per l’AR. In uno studio francese, ciò si è verificato nel 6-9% dei casi. Quando i pazienti con artrite isolata sono stati esaminati, hanno mostrato una sierologia RA positiva fino al 30% (ACPA nel 28%). Questi pazienti hanno un coinvolgimento articolare del 100%, molto probabilmente con erosioni.
I polmoni sono uno degli organi che determinano la prognosi. L’interessamento polmonare si presenta sotto forma di malattia polmonare interstiziale (ILD), cioè alveolite o fibrosi, nel 60-80% dei pazienti con anticorpi Jo-1, più frequentemente (>80%) nei pazienti con non-Jo-1-Ak. Può essere clinicamente acuta (10-30%) con esordio (per)acuto, rapida progressione con possibile deterioramento in pochi giorni fino a richiedere cure intensive o ventilazione, febbre frequente e talvolta insufficienza respiratoria acuta. Il 40-50% dei casi è cronico, con un esordio insidioso e una lenta progressione, fino a quando si sviluppano dispnea, tosse irritabile e riduzione della funzione polmonare. Il 20-25% dei pazienti è asintomatico e presenta solo anomalie nelle letture o nei risultati della TAC. Se le persone colpite sopravvivono alla fase acuta, la prognosi a lungo termine è la stessa per tutte e tre le forme di progressione. Non bisogna dimenticare che l’ipertensione polmonare (PH) può verificarsi anche nelle sindromi da antisintetasi. Questo si verifica in quasi il 10% dei casi con un Jo-1-Ak e fino al 20% dei pazienti con un non-Jo-1-Ak e può anche essere indipendente dalla stessa ILD, ad esempio come PH “sproporzionata”, a volte anche come PH primaria.
La latenza della diagnosi di PH rispetto alla diagnosi iniziale di sindrome antisintetica è molto alta. In alcuni casi, la diagnosi è stata fatta solo quando la PH era già molto avanzata. Secondo il dottor Bauhammer, questo potrebbe essere dovuto principalmente al fatto che i medici pensano alla possibilità di ipertensione polmonare troppo tardi. Di conseguenza, la prognosi è scarsa quando si verifica la PH. Il tasso di sopravvivenza a 3 anni qui è solo del 58%. Rispetto ai pazienti senza PH, quelli con ipertensione polmonare hanno una marcata riduzione della DLco/FVC e hanno maggiori probabilità di avere la sindrome di Raynaud e una microscopia capillare anormale.
I segni cutanei nelle sindromi da antisintetasi sono le cosiddette mani del meccanico o – quando i segni si manifestano sui piedi – i “piedi dell’escursionista” (Fig. 2) . Circa un terzo dei pazienti ne è affetto. Sono caratterizzate da ipercheratosi ed eritema non prurito, soprattutto sul lato radiale-palmare delle dita e sui palmi delle mani. Spesso ci sono associazioni con l’ILD (grave). Le lesioni cutanee regrediscono con la terapia sistemica, la terapia locale specifica non è quasi mai necessaria.
I sintomi secondari dell’ ASA, cioè Raynaud o febbre, sono presenti nel 30-40% dei pazienti. La sindrome di Raynaud non è generalmente così grave come la sclerodermia, ma ci sono casi di ulcere digitali e persino di ischemia digitale acuta grave. La febbre è più comune nei pazienti che presentano un coinvolgimento polmonare acuto. L’esperto ha anche riferito di uno studio che ha valutato una nuova comparsa di Raynaud, mani da meccanico o febbre nel corso della malattia come foriera di una ricaduta acuta e/o di una nuova manifestazione d’organo (rischio 4 volte superiore con Jo-1-Ak).
Terapia
La terapia dipende fortemente dal coinvolgimento dell’organo, cioè gli immunosoppressori e gli steroidi vengono scelti in base all’organo colpito e alla sua gravità. Di conseguenza, esiste anche un’ampia gamma di farmaci che possono essere utilizzati. Nella malattia polmonare interstiziale, che è un organo importante dal punto di vista prognostico, gli inibitori della calcineurina, in particolare, hanno dimostrato di essere molto efficaci e, a seconda della gravità, la ciclofosfamide o il rituximab. Ci sono anche segnalazioni di casi di buona risposta al MMF o alle immunoglobuline per via endovenosa, anche se i dati su queste ultime sono ancora molto scarsi nei casi di coinvolgimento polmonare. Gli inibitori del TNF-alfa sono stati utilizzati nel coinvolgimento articolare, ma la miosite e la malattia polmonare interstiziale possono peggiorare con questi farmaci (progressione in ≥33%). Dal 2020, in Germania esiste una nuova opzione per l’uso di nintedanib per la fibrosi polmonare progressiva, che è stato approvato per la sclerosi sistemica con coinvolgimento polmonare da marzo e per le malattie polmonari interstiziali fibrosanti croniche e progressive di altro tipo da luglio e quindi potenzialmente anche per i pazienti con antisintetasi. Secondo il consiglio del dottor Bauhammer, bisogna sempre tenere conto di una buona terapia di accompagnamento (ginnastica respiratoria, fisioterapia).
Previsioni
Il decorso della miosite è generalmente considerato più lieve rispetto, ad esempio, al decorso della miosite con anticorpi SRP. I pazienti che hanno un coinvolgimento polmonare hanno in genere una mortalità più elevata rispetto ai pazienti che non ne hanno. I soggetti affetti da coinvolgimento polmonare in una ASD – anche se si sviluppa una fibrosi – hanno una sopravvivenza significativamente migliore rispetto ai pazienti con fibrosi polmonare idiopatica, anche con un modello UIP sulla diagnostica per immagini o sull’istologia.
Messaggi da portare a casa
- Ricordiamo che il paziente può avere un esordio ASA, spesso mono/oligosintomatico.
- Indicativi di ASA nell’artrite isolata: sindrome di Raynaud, anti-Ro52-Ak e/o mani/piedi da meccanico, tra gli altri.
- La nuova comparsa di Raynaud, di segni cutanei e di febbre può preannunciare una ricaduta o una nuova manifestazione d’organo.
- La scelta dell’immunosoppressione dipende dal coinvolgimento degli organi, dimostrato nella ILD: Inibitori della calcineurina, RIX
- Non dimentichi lo screening del PH
- Previsioni relativamente buone
– Aggiornamento FomF Rheuma Nephro (online), 29-31.10.2020
Fonte:
- Conferenza “Sindromi antisintetiche“ all’aggiornamento FomF Rheuma Nephro (online), 29.10.2020
InFo PAIN & GERIATURE 2020; 2(2): 42-43 (pubblicato il 10.12.20, prima della stampa).