
Oltre alle cure standard per tutte le forme di SLA, ci sono stati progressi significativi, in particolare per le forme monogenetiche di SLA, grazie allo sviluppo di metodi di terapia specifici basati sui geni, attualmente l’ASO in particolare. La diagnostica genetica di base, almeno dei geni più comuni (SOD1, C9orf72, FUS, TARDP), è quindi raccomandata per tutti i pazienti con SLA al momento della diagnosi.
Può sostenere il test ECM nella nostra piattaforma di apprendimento dopo aver esaminato i materiali consigliati. Clicchi sul seguente pulsante:
I casi di sclerosi laterale amiotrofica (SLA) sono stati descritti per la prima volta da Jean Martin Charcot nel 1873 [1]. Charcot aveva già fornito prove neuropatologiche di una degenerazione del sistema motorio sottostante. Oggi è noto che la sclerosi laterale amiotrofica è una neurodegenerazione multisistemica con numerose manifestazioni extramotorie, nonostante la degenerazione predominante del primo e del secondo motoneurone e del tratto corticospinale. Abbiamo acquisito una conoscenza sempre maggiore dei fattori genetici sottostanti, in particolare nell’ultimo decennio, che ha portato anche alle prime conseguenze terapeutiche dirette.
Epidemiologia
Sulla base dei dati del registro dei pazienti meglio gestito in Svevia, si può stimare che ci siano circa 8.000-9.000 persone affette da SLA nell’intera Germania [2]. Con un’età media di insorgenza di 70-75 anni e una leggera predominanza maschile, si ipotizza un’incidenza di circa 3/100.000 pazienti. La prevalenza nell’arco della vita, come misura statistica più descrittiva per la probabilità di sviluppare la SLA, è di 1:400. Questi dati epidemiologici mostrano una grande congruenza con i dati di altri Paesi europei. [3,4]In altre parti del mondo, come l’Asia, i dati epidemiologici sono diversi.
Sintomi clinici
I sintomi principali della SLA sono una paresi atrofica ad esordio focale, progressiva, con frequenti spasmi muscolari e fascicolazioni, come segno di un danno crescente ai secondi motoneuroni a livello spinale o nell’area del tronco encefalico [5]. Questo è preceduto o accompagnato da un’affezione dei primi motoneuroni nella corteccia motoria primaria e nel tratto corticospinale, con riflessi di stiramento muscolare aumentati e saltati ed evidenze di riflessi patologici o un aumento del tono muscolare nel senso di spasticità.
[6,7]I deficit cognitivi e le anomalie comportamentali rilevanti dal punto di vista clinico e della vita quotidiana, nel senso di una sindrome di demenza frontotemporale, sono riscontrati come sintomi extra-motori in circa il 5% delle persone affette da SLA. Negli ultimi anni sono stati descritti sempre più spesso disturbi di accompagnamento del sistema nervoso autonomo [8]. Inoltre, anche il dolore di varia origine è importante durante il decorso della malattia [9].Procedure diagnostiche e diagnosi
La diagnosi viene fatta principalmente a livello clinico, dopo l’esclusione coerente e attenta di diagnosi differenziali alternative. [10]I criteri di Gold Coast forniscono una buona guida per la diagnosi clinica. La procedura diagnostica di supporto più importante è l’elettromiografia, per rilevare la denervazione attiva o i potenziali di fascicolazione polipodica con la contemporanea evidenza di un danno neurogenico cronico. [11]Nell’elettromiografia, devono essere esaminati diversi muscoli di tutte e quattro le regioni del corpo (nervi cranici/bulbari – cervicali – toracici – lombosacrali). Oltre all’elettromiografia, si può eseguire anche la sonografia muscolare per determinare la presenza di fascicolazioni muscolari polipoidi e per valutare il trofismo muscolare e la struttura interna. L’ecografia muscolare può essere particolarmente vantaggiosa per il rilevamento di fascicolazioni muscolari in muscoli più grandi o più profondi, come parti del muscolo quadricipite femorale o dei muscoli della base della lingua, e può essere un complemento importante all’elettromiografia. L’elettroneurografia dei nervi motori e sensoriali e la diagnostica delle onde F degli arti superiori e inferiori sono particolarmente necessarie per escludere la neuropatia demielinizzante primaria e, in particolare, i blocchi di conduzione (CIDP o neuropatia motoria multifocale/MMN).
La stimolazione magnetica transcranica per la registrazione dei potenziali evocati dal motore è disponibile per oggettivare e, se necessario, quantificare il coinvolgimento dei primi motoneuroni. [12]Questo metodo di esame può essere utilizzato per esaminare la conduzione motoria centrale degli assoni densamente mielinizzati del tratto corticospinale dalla corteccia motoria come sito di stimolazione lungo tutto il midollo spinale. È opportuno esaminare un muscolo distale ciascuno degli arti superiori e inferiori. La Tabella 1 fornisce una panoramica dettagliata delle diagnosi elettrofisiologiche proposte per la SLA e dei criteri diagnostici della Gold Coast .
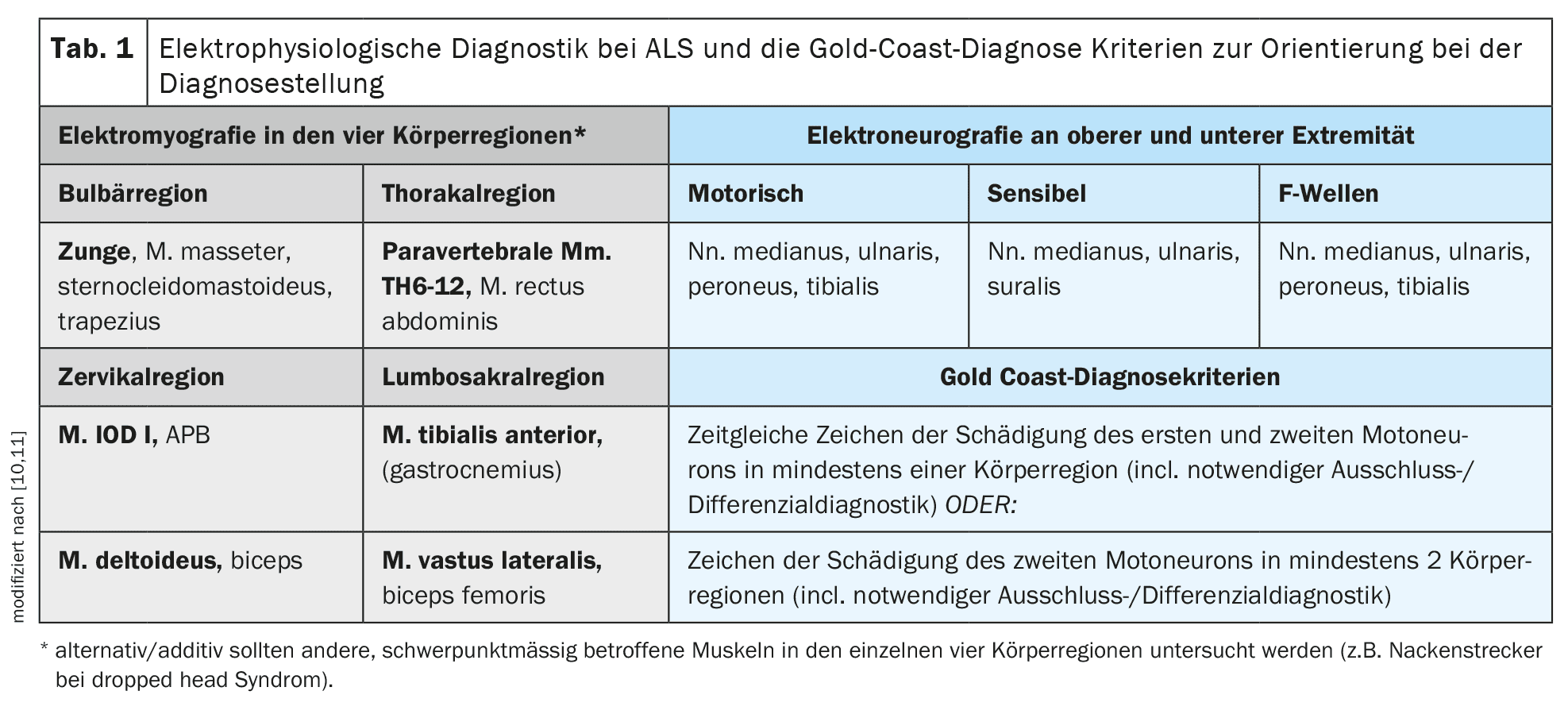
Mentre la diagnostica genetica era ancora considerata facoltativa nella linea guida S1, valida e disponibile dal 2021, gli autori ritengono che questa situazione sia cambiata in seguito agli sviluppi degli ultimi tre anni [5]. [15,16]In vista degli sviluppi terapeutici in questo campo, ogni paziente affetto da SLA dovrebbe sottoporsi obbligatoriamente a test genetici mirati, almeno per la presenza di una mutazione nel gene della superossido dismutasi 1 Cu/Zn(SOD 1) e i giovani pazienti affetti da SLA di età inferiore ai 40 anni per la presenza di una mutazione patogena nel gene FUS. [17]Opzionalmente, dovrebbe essere effettuata anche una diagnostica genetica più estesa con l’esame di altri geni come C9orf72, TARDP, TBK1 ecc. Le sezioni seguenti sull’eziologia e la genetica e, in particolare, sulla terapia tratteranno questi temi in modo più dettagliato. La Tabella 1 fornisce una panoramica delle procedure diagnostiche descritte e dei criteri diagnostici Gold Coast raccomandati come guida nella pratica clinica .
Per quanto riguarda ulteriori indagini clinicamente rilevanti per una diagnosi differenziale e di esclusione completa, in base alla presentazione clinica iniziale (ad esempio, risonanza magnetica per immagini, diagnostica otorinolaringoiatrica, diagnostica di laboratorio), così come per la diagnostica rilevante durante il decorso della malattia per valutare la prognosi (ad esempio, test di funzionalità polmonare/test di funzionalità diaframmatica, diagnostica della deglutizione con FEES, psicometria con ECAS), vorremmo fare esplicito riferimento alla presentazione molto dettagliata e chiara della linea guida S1 [5].
Spettro fenotipico e modello di paresi muscolare sullo sfondo della neuroanatomia e della fisiopatologia della SLA
Le scoperte scientifiche degli ultimi due decenni, in particolare gli studi neuroanatomici e neuropatologici, hanno cambiato radicalmente la visione della SLA. [18,19]Oggi, la SLA non è più vista come una degenerazione puramente del sistema motorio, ma come una degenerazione multisistemica.
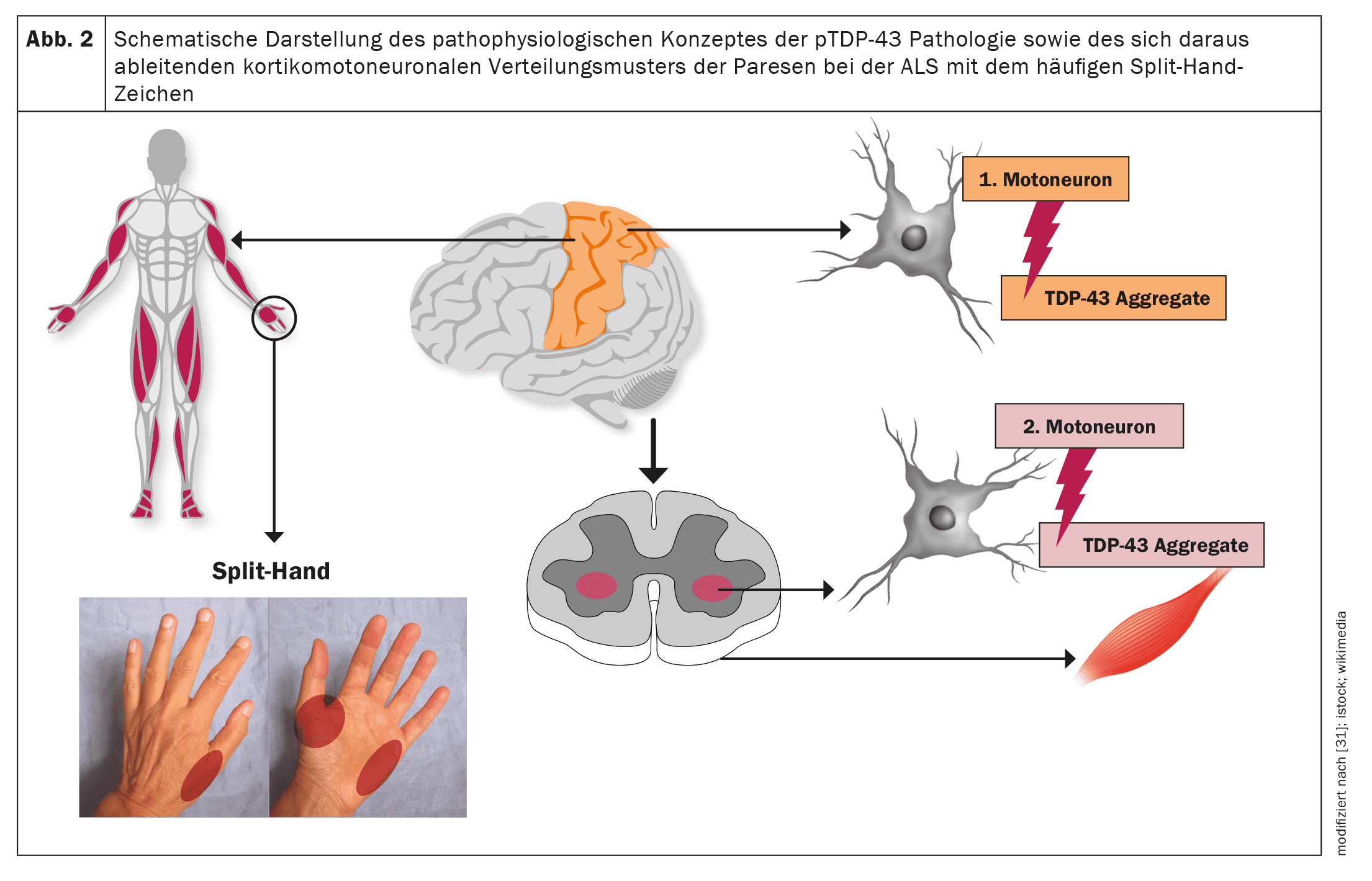
La SLA, come altre malattie neurodegenerative come il morbo di Alzheimer o il morbo di Parkinson, è una proteinopatia, cioè gli aggregati proteici patologici nelle cellule nervose motorie, come correlazione neuropatologica, portano alla disfunzione delle cellule nervose motorie colpite e, in ultima analisi, all’apoptosi e quindi alla perdita delle cellule nervose motorie. [20]Nel caso della SLA, oltre il 95% dei casi coinvolge gli aggregati proteici TDP-43. [21]Solo nel caso di varianti SOD1 o FUSpatologiche sottostanti, si verifica una neurodegenerazione indipendente dalla TDP-43. [22]Gli studi neuropatologici hanno stabilito una propagazione cerebrale graduale e a tappe della patologia TDP-43 nella SLA, paragonabile alla patologia da α-sinucleina nella malattia di Parkinson e alla patologia da tau nella malattia di Alzheimer. Questi risultati, così come il modello corticomotoneuronale della paresi con il segno della mano divisa (atrofia asimmetrica dei muscoli della mano C8/T1 o [23–26] come segno clinico molto comune nella SLA, implicano un’origine dei cambiamenti neuropatologici nell’area della corteccia motoria primaria e una diffusione graduale della patologia TDP-43 da lì attraverso il trasporto assonale ai secondi neuroni motori, cioè i nuclei dei nervi cranici motori nel tronco cerebrale e le cellule del corno anteriore del midollo spinale.
[27,28]Sembra che si verifichi una propagazione di tipo prionico dei cambiamenti neuropatologici, che spiegherebbe l’esordio focale della manifestazione motoria e la diffusione graduale ai miotomi e alle regioni corporee vicine. [29]Di conseguenza, a seconda della manifestazione iniziale della neuropatologia, lo spettro fenotipico della SLA – per quanto riguarda i soli sintomi motori – non è uniforme, ma altamente variabile. Ad esempio, possono verificarsi fenotipi motori topograficamente distinti. Inoltre, il fenotipo è influenzato dalla velocità di degenerazione, solitamente diversa, del primo e del secondo motoneurone e dai sintomi clinici associati. Senza una comprensione precisa dei meccanismi molecolari, il rapporto tra gli aggregati insolubili di proteina TDP-43, che non possono più essere trasportati a livello assonale e quindi si accumulano localmente, e gli oligomeri solubili di TDP-43 come precursori degli aggregati TDP-43 sembra essere decisivo. [23]Ovviamente, questi precursori solubili vengono poi trasportati attraverso il trasporto assonale dai primi motoneuroni ai secondi motoneuroni, dove si depositano obbligatoriamente come aggregati proteici e portano alla morte di queste cellule.Ad oggi, non esiste una classificazione standardizzata dei fenotipi clinici che tenga conto del rapporto o del focus del danno al primo e al secondo motoneurone, del modello clinico o del focus della paresi atrofica e della sua diffusione.
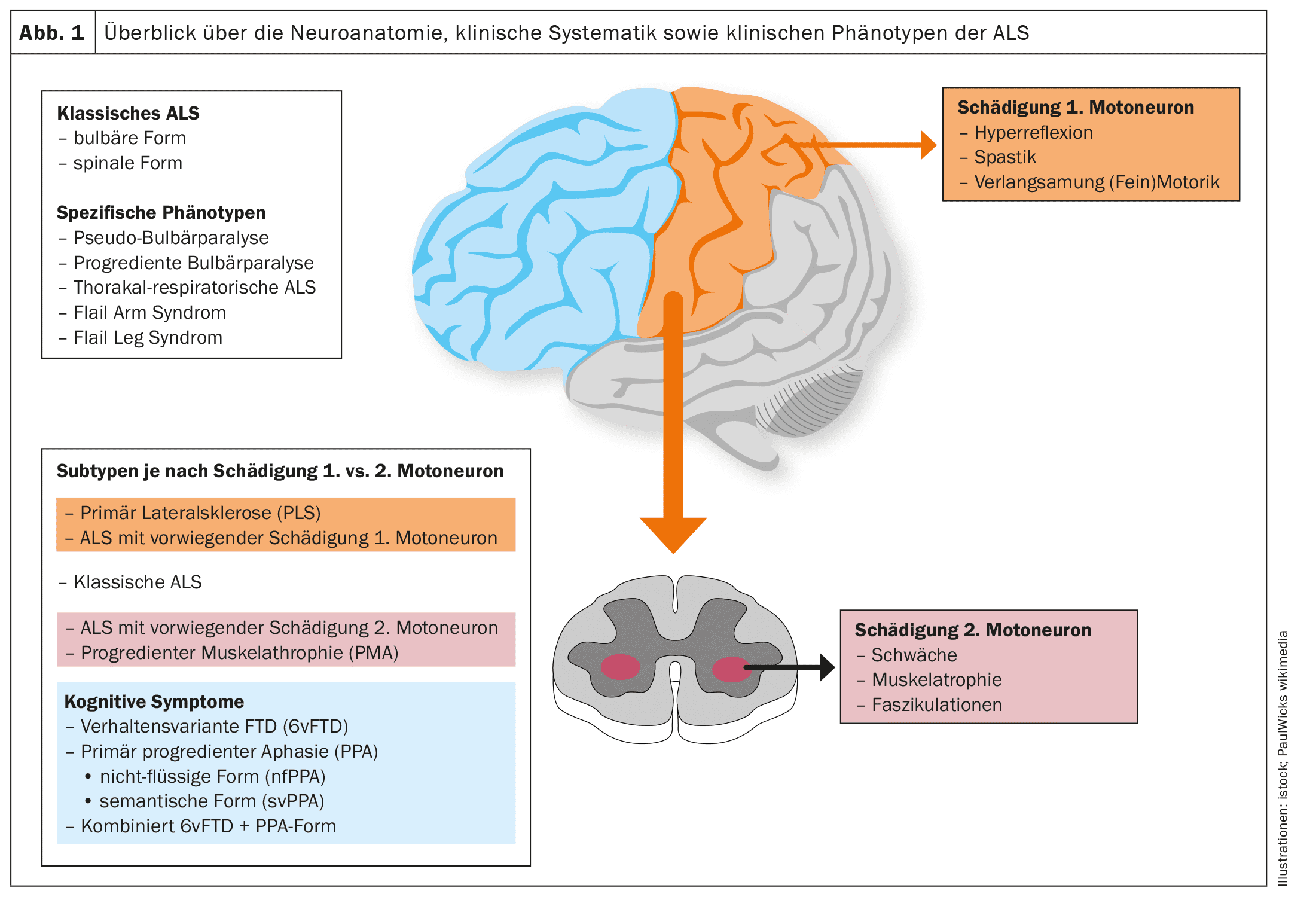
Le figure 1 e 2 forniscono una panoramica schematica della neuroanatomia e della fisiopatologia della malattia, nonché dei fenotipi clinici e della loro classificazione.
Eziologia e genetica
L’eziologia della SLA non è ancora sufficientemente ben compresa. Nella forma sporadica della malattia, che rappresenta la grande maggioranza dei casi, si ipotizza una genesi multifattoriale con l’interazione di vari fattori ambientali esterni sfavorevoli, fattori metabolici epigenetici e una possibile suscettibilità genetica aggiuntiva. [31]I cambiamenti nel trasporto di proteine nucleari-citosoliche, nel metabolismo dell’RNA, nella funzione ossidativa mitocondriale delle cellule, nell’eccitabilità glutammatergica, nel trasporto di proteine assonali e nell’autofagia cellulare sono discussi come meccanismi metabolici intracellulari sottostanti, mentre i disturbi funzionali degli atrociti e degli oligodendrociti e i processi infiammatori sono discussi come meccanismi extracellulari additivi. Le intuizioni significative sui possibili meccanismi eziologici della SLA sporadica sono state acquisite principalmente attraverso forme genetiche con processi fisiopatologici distinti.
[32]Nel 1993, sono state descritte per la prima volta mutazioni patogene nella superossido dismutasi 1 Cu/Zn (SOD1) come causa genetica della SLA. Oggi è noto che un gran numero di mutazioni genetiche sono alla base dell’eziologia della SLA. [17]Nel caso di SLA apparentemente sporadica senza una storia familiare evidente in Germania, una causa monogenetica può essere individuata in poco più del 10% dei casi. [17]Le cause genetiche più comuni in Germania sono le espansioni patologiche della ripetizione C9orf72 e le mutazioni nei geni SOD1, TARDP e FUS.La diagnosi genetica al momento della diagnosi dovrebbe quindi essere offerta a tutti i pazienti con SLA, in quanto può avere conseguenze terapeutiche immediate, che discuteremo in dettaglio nella sezione seguente.
Terapia e prognosi
Il Riluzolo è stato approvato in Germania dal 1996 come l’unica sostanza farmacologica con un effetto positivo dimostrato sulla progressione della SLA. Si ipotizza che il meccanismo d’azione decisivo sia una riduzione del rilascio di glutammato e quindi una riduzione dell’eccitotossicità. [18]Questa ipotesi è stata resa più concreta dall’evidenza della degenerazione primaria delle vie glutammatergiche corticofughe. Una dose giornaliera di riluzolo di 100 mg ha il miglior profilo efficacia/effetti collaterali. [33–35]Le analisi retrospettive di un totale di dieci registri clinici sulla SLA forniscono prove di un prolungamento mediano del tempo di sopravvivenza fino a 19 mesi con il riluzolo, e ci sono anche chiare indicazioni che questa sostanza è efficace anche nelle fasi più avanzate della malattia. Il riluzolo è generalmente ben tollerato; gli effetti collaterali noti includono un aumento delle transaminasi, che devono quindi essere monitorate regolarmente, e disturbi gastrointestinali. Per i pazienti affetti da SLA con disfagia sono ora disponibili anche forme di dosaggio alternative, come il succo o le compresse orodispersibili.
Oltre alla terapia farmacologica con riluzolo, si ritiene che la prevenzione di uno stato metabolico catabolico con una perdita di peso consecutiva abbia un ulteriore beneficio prognostico. [36]Più alto è l’indice di massa corporea (BMI), più favorevole è la prognosi.
Le raccomandazioni attuali per i pazienti affetti da SLA sono quindi di mantenere un peso stabile e di evitare la perdita di peso. [37]In questo contesto, anche l’inserimento di un tubo PEG svolge un ruolo importante nella disfagia progressiva, con un prolungamento del tempo di sopravvivenza come risultato di questa misura. Se e in che misura gli approcci terapeutici anticatabolici specifici, come gli interventi nutrizionali mirati ad alto contenuto di grassi e di calorie o gli interventi nutrizionali chetogenici, possano migliorare la prognosi, sono attualmente oggetto di studi intensivi. In questo contesto, lo studio LIPCAL-ALS 2, condotto in Germania come Investigator Initiated Trial (IIT) dai colleghi di Ulm, il cui inizio è previsto tra il 2024 e il 2025, è di grande importanza.
[38,39]La ventilazione non invasiva (NIV) e invasiva per mezzo di un tracheostoma è un’altra misura importante per prolungare il tempo di sopravvivenza nella SLA con insufficienza ventilatoria. Questo è anche plausibile, in quanto l’ipoventilazione alveolare con ipercapnia consecutiva dovuta al coinvolgimento diaframmatico è tipicamente un fattore significativo nella morte dei pazienti con SLA dopo tre-cinque anni di progressione della malattia.Lo sviluppo di oligonucleotidi antisenso (ASO) per specifiche forme geneticamente indotte di SLA può essere considerato una pietra miliare. In particolare, va menzionato l’ASO Tofersen somministrato per via intratecale, che lega l’ mRNA della SOD1nei pazienti con variantipatogene della SOD1(circa l’1-2% di tutti i casi di SLA) e quindi impedisce l’espressione citotossica della proteina SOD1. [16,40]È stato dimostrato che l’effetto meccanicistico di una riduzione significativa dell’espressione della SOD1di circa il 30% negli esseri umani si verifica molto rapidamente entro pochi giorni dall’inizio della terapia con Tofersen, seguita da un calo significativo della NfL nel liquor e nel siero, prima che si verifichi infine un rallentamento del declino del punteggio ALSFRS-R. Dopo un’osservazione a lungo termine di dodici mesi, sono stati osservati ulteriori segnali positivi con rilevanza clinica, come gli effetti sulla funzione muscolare e la stabilizzazione del peso.
Negli Stati Uniti, Tofersen è stato approvato dalla FDA nell’aprile 2023 esclusivamente sulla base dei dati convincenti dei biomarcatori, con un calo significativo dei valori di NfL. In Germania, Tofersen è stato reso disponibile come parte di un programma di accesso aperto. [41]I dati iniziali di applicazione nel mondo reale della rete tedesca per il motoneurone hanno confermato in modo impressionante i dati dello studio di Tofersen, con dati ancora migliori in termini di parametri di progressione clinica. In questo contesto, era logico che l’EMA decidesse a favore dell’approvazione di Tofersen nel febbraio 2024.
[15,42]Oltre all’ASO Tofersen per la SLA associata a SOD1, sono attualmente in fase di sviluppo o sono già stati esaminati in studi, in particolare contro C9orf72 e FUS . In particolare, va menzionato l’ASO Jacifusen (ION363) quando viene individuata unavariante patogena FUS come causa della SLA giovanile, che è attualmente in fase di sperimentazione in uno studio multicentrico e multinazionale con due siti di studio (Rostock e Ulm) in Germania. [43]La Scala di Valutazione Funzionale della SLA rivista (ALSFRS-R) è un punteggio collaudato e consolidato per valutare le funzioni motorie di tutte e quattro le regioni del corpo e non solo è un importante endpoint per gli studi clinici, ma ha anche dimostrato di essere un parametro di follow-up facilmente realizzabile nella pratica clinica. [44]L’ALSFRS-SE (SE: “autoesplicativo”), che è stato concordato solo di recente all’interno della rete tedesca MND in lingua tedesca, con le relative spiegazioni concrete ed esemplari per i singoli item e le menomazioni funzionali, dovrebbe avere un ulteriore vantaggio in termini di gestione pratica e, in particolare, di accuratezza diagnostica.Conclusione per la pratica
Oltre alle cure standard per tutte le forme di SLA (Riluzolo, prevenzione dello stato metabolico catabolico, compreso l’inserimento tempestivo di un tubo PEG, ventilazione precoce non invasiva e, se necessario, invasiva in caso di insufficienza ventilatoria), si sono registrati progressi significativi, in particolare per le forme monogenetiche di SLA, grazie allo sviluppo di metodi terapeutici specifici basati sui geni, attualmente in particolare l’ASO. A questo proposito, va menzionato Tofersen, una terapia molto efficace e specifica per la SLA associata a SOD1, che ora è disponibile anche in Germania.
La diagnostica genetica di base, almeno dei geni più comuni (SOD1, C9orf72, FUS, TARDP), è quindi raccomandata per tutti i pazienti con SLA al momento della diagnosi. La misura in cui gli interventi mirati, anticatabolici e ipercalorici possono influenzare favorevolmente il decorso della SLA sporadica sarà mostrata, si spera, dai prossimi studi in questo settore.
Messaggi da portare a casa
- Riluzolo 100 mg/d, la prevenzione di uno stato metabolico catabolico, compreso l’inserimento tempestivo di un tubo PEG e la ventilazione precoce non invasiva in caso di insufficienza respiratoria, sono disponibili come cura standard per tutte le forme di SLA. L’inserimento tempestivo di un tubo PEG, così come la ventilazione precoce non invasiva e, se necessario, invasiva per l’insufficienza ventilatoria.
- Inoltre, sono stati compiuti progressi significativi, in particolare per le forme monogenetiche di SLA. La base di questo è lo sviluppo di metodi terapeutici specifici basati sui geni, come gli oligonucleotidi antisenso (ASO) in particolare. Il Tofersen deve essere menzionato come una terapia molto efficace e specifica per la SLA associata a SOD1.
- La diagnostica genetica di base, almeno dei geni più comuni (SOD1, C9orf72, FUS, TARDP) è quindi raccomandata per tutti i pazienti con SLA al momento della diagnosi.
Letteratura:
- Duyckaerts C, Maisonobe T, Hauw JJ, Seilhean D: Charcot identifica e illustra la sclerosi laterale amiotrofica. Free Neuropathol. 2. doi:10.17879/freeneuropathology-2021-3323.
- Uenal H, Rosenbohm A, Kufeldt J, et al: Incidenza e variazione geografica della Sclerosi Laterale Amiotrofica (SLA) nella Germania meridionale – Completezza del Registro SLA Swabia. PLoS ONE. 2014; 9(4). doi:10.1371/journal.pone.0093932.
- Jun KY, Park J, Oh KW, et al: Epidemiologia della SLA in Corea utilizzando i big data nazionali. J Neurol Neurosurg Psychiatry. 2019;90(4): 395-403. doi:10.1136/jnnp-2018-318974.
- Marin B, Boumédiene F, Logroscino G, et al: Variazione dell’incidenza mondiale della sclerosi laterale amiotrofica: una meta-analisi. Int J Epidemiol 2017; 46(1): 57-74. doi:10.1093/ije/dyw061.
- Petri SA-O GJ Ludolph AC. “Malattie del motoneurone” della Società tedesca di neurologia (DGN). (2524-2348; Elettronico).
- Finsel J, Uttner I, Vázquez Medrano CR, et al: La cognizione nel corso della SLA – una meta-analisi. Amyotroph Lateral Scler Front Degener. 2023; 24(1-2): 2-13. doi:10.1080/21678421.2022.2101379.
- Iazzolino B, Pain D, Peotta L, et al: Validazione della classificazione rivista dei disturbi cognitivi e comportamentali nella SLA. J Neurol Neurosurg Psychiatry 2019; 90(7): 734-739. doi:10.1136/jnnp-2018-319696.
- Oprisan AL, Popescu BO: Disautonomia nella Sclerosi Laterale Amiotrofica. Int J Mol Sci 2023; 24(19). doi:10.3390/ijms241914927.
- Chiò A, Mora G, Lauria G. Il dolore nella sclerosi laterale amiotrofica. Lancet Neurol 2017; 16(2): 144-157. doi:10.1016/S1474-4422(16)30358-1
- Shefner JM, Al-Chalabi A, Baker MR, et al: Una proposta di nuovi criteri diagnostici per la SLA. Clin Neurophysiol 2020; 131(8): 1975-1978. doi:10.1016/j.clinph.2020.04.005.
- Koch JC, Petri S, Zeller D: Diagnosi elettrofisiologica del sospetto di sclerosi laterale amiotrofica – Raccomandazioni di consenso della Rete tedesca per le malattie del motoneurone (MND-NET). Clin Neurophysiol 2024. 2024; 55: 82-88.
- Zoccolella S, Mastronardi A, Scarafino A, et al: Potenziali evocati motori nella sclerosi laterale amiotrofica: potenziali implicazioni nel rilevare il coinvolgimento subclinico dell’UMN nel fenotipo del motoneurone inferiore. J Neurol 2020; 267(12): 3689-3695. doi:10.1007/s00415-020-10073-5.
- Shahim P, Norato G, Sinaii N, et al. Neurofilamenti nella Sclerosi Laterale Amiotrofica sporadica e familiare: una revisione sistematica e una meta-analisi. Genes. 2024;15(4). doi:10.3390/genes15040496.
- Behzadi A, Pujol-Calderón F, Tjust AE, et al: I neurofilamenti possono differenziare i sottogruppi di SLA e la SLA dai comuni mimici diagnostici. Sci Rep 2021; 11. doi:10.1038/s41598-021-01499-6.
- Korobeynikov VA, Lyashchenko AK, Blanco-Redondo B, et al: Silenziamento con oligonucleotidi antisenso dell’espressione di FUS come approccio terapeutico nella sclerosi laterale amiotrofica. Nat Med 2022; 28(1): 104-116. doi:10.1038/s41591-021-01615-z.
- Miller TM, Cudkowicz ME, Genge A, et al: Prova dell’oligonucleotide antisenso Tofersen per la SLA SOD1. N Engl J Med. Pubblicato online il 22 settembre 2022. doi:10.1056/NEJMoa2204705.
- Ruf WP, Boros M, Freischmidt A, et al: Spettro e frequenza delle varianti genetiche nella sclerosi laterale amiotrofica sporadica. Brain Commun 2023; 5(3). doi:10.1093/braincomms/fcad152.
- Braak H, Brettschneider J, Ludolph AC, et al: Sclerosi laterale amiotrofica – un modello di diffusione assonale corticofuga. Nat Rev Neurol 2013; 9(12): 708-714. doi:10.1038/nrneurol.2013.221.
- Brettschneider J, Del Tredici K, Toledo JB, et al: Fasi della patologia pTDP-43 nella sclerosi laterale amiotrofica. Ann Neurol 2013; 74(1): 20-38. doi:10.1002/ana.23937.
- Neumann M: Neuropatologia molecolare delle proteinopatie TDP-43. Int J Mol Sci 2009; 10(1): 232-246. doi:10.3390/ijms10010232.
- Saberi S, Stauffer JE, Schulte DJ, Ravits J: “Neuropatologia della sclerosi laterale amiotrofica e delle sue varianti”. Neurol Clin 2015; 33(4): 855-876. doi:10.1016/j.ncl.2015.07.012.
- Braak H, Braak E: Stadiazione dei cambiamenti neurofibrillari legati alla malattia di Alzheimer. Neurobiol Aging 1995; 16(3): 271-278-284.
- Braak H, Ludolph AC: Neumann M, Ravits J, Del Tredici K. I cambiamenti patologici TDP-43 nelle cellule di Betz differiscono da quelli negli α-motoneuroni bulbari e spinali nella sclerosi laterale amiotrofica sporadica.
Acta Neuropathol (Berl) 2017; 133(1): 79-90. doi:10.1007/s00401-016-1633-2. - Eisen A, Braak H, Del Tredici K, et al: Le influenze corticali guidano la sclerosi laterale amiotrofica. J Neurol Neurosurg Psychiatry 2017; 88(11): 917-924. doi:10.1136/jnnp-2017-315573.
- Ludolph AC, Emilian S, Dreyhaupt J, et al: Il modello di paresi nella SLA è coerente con la fisiologia delle proiezioni corticomotoneuronali ai diversi gruppi muscolari. J Neurol Neurosurg Psychiatry 2020; 91(9): 991-998. doi:10.1136/jnnp-2020-323331.
- Menon P, Kiernan MC, Vucic S: L’ipereccitabilità corticale precede la disfunzione del motoneurone inferiore nella SLA. Clin Neurophysiol 2015; 126(4): 803-809. doi:10.1016/j.clinph.2014.04.023.
- Prasad A, Bharathi V, Sivalingam V, et al: Meccanismi molecolari del dispiegamento di TDP-43 e patologia nella Sclerosi Laterale Amiotrofica. Front Mol Neurosci 2019;12. doi:10.3389/fnmol.2019.00025.
- Gosset P, Camu W, Raoul C, Mezghrani A: Prionoidi nella sclerosi laterale amiotrofica. Brain Commun 2022;4(3). doi:10.1093/braincomms/fcac145.
- Hardiman O, Al-Chalabi A, Chio A, et al: Sclerosi laterale amiotrofica. Nat Rev Dis Primer 2017; 3:17071. doi:10.1038/nrdp.2017.71.
- Masrori P, Van Damme P: Sclerosi laterale amiotrofica: una revisione clinica. Eur J Neurol 2020; 27(10): 1918-1929. doi:10.1111/ene.14393.
- Eisen A, Vucic S, Mitsumoto H: Storia della SLA e teorie concorrenti sulla patogenesi: capitolo del manuale IFCN. Clin Neurophysiol Practice 2023; 9: 1-12. doi:10.1016/j.cnp.2023.11.004.
- Rosen DR, Siddique T, Patterson D, et al: Mutazioni nel gene della superossido dismutasi Cu/Zn sono associate alla sclerosi laterale amiotrofica familiare. Nature 1993; 362(6415): 59-62. doi:10.1038/362059a0.
- Bensimon G, Lacomblez L, Meininger V, Gruppo AS: Uno studio controllato di Riluzolo nella Sclerosi Laterale Amiotrofica. http://dx.doi.org/10.1056/NEJM199403033300901.
- Hinchcliffe M, Smith A: Riluzolo: le prove del mondo reale supportano un significativo prolungamento dei tempi di sopravvivenza mediana nei pazienti con sclerosi laterale amiotrofica. Degener Neurol Neuromuscul Dis 2017;7: 61-70. doi:10.2147/DNND.S135748.
- Miller RG, Mitchell JD, Moore DH: Riluzolo per la sclerosi laterale amiotrofica (SLA)/malattia del motoneurone (MND).
Cochrane Database Syst Rev 2012; 2012(3).
doi:10.1002/14651858.CD001447.pub3. - Dupuis L, Pradat PF, Ludolph AC, Loeffler JP: Metabolismo energetico nella sclerosi laterale amiotrofica. Lancet Neurol 2011; 10(1): 75-82. doi:10.1016/S1474-4422(10)70224-6.
- Cui F, Sun L, Xiong J, et al: Effetti terapeutici della gastrostomia endoscopica percutanea sulla sopravvivenza nei pazienti con sclerosi laterale amiotrofica: una meta-analisi. PLoS ONE 2018; 13(2). doi:10.1371/journal.pone.0192243.
- Dorst J, Ludolph AC: Ventilazione non invasiva nella sclerosi laterale amiotrofica. Ther Adv Neurol Disord 2019; 12: 1756286419857040. doi:10.1177/1756286419857040.
- Radunovic A, Annane D, Rafiq MK, et al: Ventilazione meccanica per la sclerosi laterale amiotrofica/malattia del motoneurone.
Cochrane Database Syst Rev 2017; 2017(10). doi:10.1002/14651858.CD004427.pub4. - Miller T, Cudkowicz M, Shaw PJ, et al: Studio di fase 1-2 dell’oligonucleotide antisenso Tofersen per la SLA SOD1. N Engl J Med 2020; 383(2): 109-119. doi:10.1056/NEJMoa2003715.
- Wiesenfarth M, Dorst J, Brenner D, et al: Effetti del trattamento con tofersen nei pazienti con SOD1-ALS in un contesto “real-world” – uno studio di coorte multicentrico di 12 mesi del programma tedesco di accesso anticipato. eClinicalMedicine. 2024; 69. doi:10.1016/j.eclinm.2024.102495.
- Meijboom KE, Brown RH: Approcci alla terapia di modulazione genica per la SLA. Neurotherapeutics 2022;19(4): 1159-1179. doi:10.1007/s13311-022-01285-w.
- Cedarbaum JM, Stambler N, Malta E, et al: L’ALSFRS-R: una scala di valutazione funzionale della SLA rivista che incorpora valutazioni della funzione respiratoria. Gruppo di studio BDNF sulla SLA (Fase III). J Neurol Sci 1999; 169(1-2): 13-21. doi:10.1016/s0022-510x(99)00210-5.
- Maier A, Boentert M, Reilich P, et al: ALSFRS-R-SE: una versione adattata, annotata e autoesplicativa della scala di valutazione funzionale della sclerosi laterale amiotrofica rivista. Neurol Res Pract 2022; 4. doi:10.1186/s42466-022-00224-6.
| Pubblicato per la prima volta in neuro aktuell 2024; 38(8): 30-35. |
InFo NEUROLOGIA & PSICHIATRIA 2024; 22(4): 6-11









