Gli antistaminici H1 si dividono in una prima generazione, più vecchia, con effetti collaterali sedativi e una seconda generazione senza questi effetti. Il nostro autore fa luce sulla farmacologia di questo gruppo di principi attivi.
Finora sono noti quattro diversi recettori dell’istamina. Queste molecole accoppiate a proteine G si trovano sulla superficie cellulare ed esercitano effetti diversi a seconda del sito di espressione. Mentre l’attivazione del recettore H1 porta in particolare al prurito, alla vasodilatazione, alla contrazione della muscolatura liscia con broncospasmo o crampi addominali, alla secrezione di muco con rinorrea e all’aumento della secrezione bronchiale, nonché all’aumento della permeabilità vascolare, i recettori H2 sono coinvolti in particolare nell’aumento del succo gastrico e della secrezione acida. Inoltre, ci sono i recettori H3, che svolgono un ruolo nel SNC come autorecettori presinaptici, e i recettori H4, che svolgono un ruolo nella differenziazione e nella modulazione delle cellule immunitarie, tra le altre cose. In questo articolo, vengono discusse le sostanze dirette contro il recettore H1, mentre gli antagonisti del recettore H2, di cui solo la ranitidina è ancora in commercio in Svizzera, non vengono trattati. Agonisti e antagonisti dei recettori H3 e H4 sono in fase di sviluppo clinico.
Farmacodinamica degli antistaminici H1
In Svizzera sono disponibili 22 principi attivi della classe degli antistaminici H1. Oggi si presume che gli antistaminici stabilizzino il recettore H1 nella sua conformazione inattiva e quindi garantiscano che un minor numero di recettori possa essere attivato dall’istamina. Mentre gli antistaminici H1 della prima generazione, più vecchia, penetrano bene nel sistema nervoso centrale ed esercitano un effetto sedativo sui recettori H1 postsinaptici, questo non avviene con i rappresentanti della seconda generazione o non in concentrazioni terapeutiche (Tab. 1) . Grazie alla loro buona efficacia nei confronti dei recettori H1 centrali, alcuni rappresentanti del gruppo Sedativi/ipnotici di prima generazione (es. doxilamina, difenidramina), antiemetici (es. meclozina) o contro la cinetosi (es. dimenidrinato). La scarsa penetrazione nel sistema nervoso centrale dei rappresentanti del La seconda generazione è dovuta al fatto che queste sostanze sono idrofile e substrati del trasportatore P-glicoproteina diretto verso l’esterno, presente nella barriera emato-encefalica (tra le altre barriere di membrana del corpo). In questo modo si evita la sedazione che si verifica nelle indicazioni antiallergiche con sostanze del tipo La prima generazione era spesso limitante per la terapia. Alcuni antistaminici H1 del tipo La prima generazione ha effetti aggiuntivi sui recettori dell’acetilcolina, della noradrenalina e della serotonina, mentre i rappresentanti della La seconda generazione inattiva in modo specifico il recettore H1.
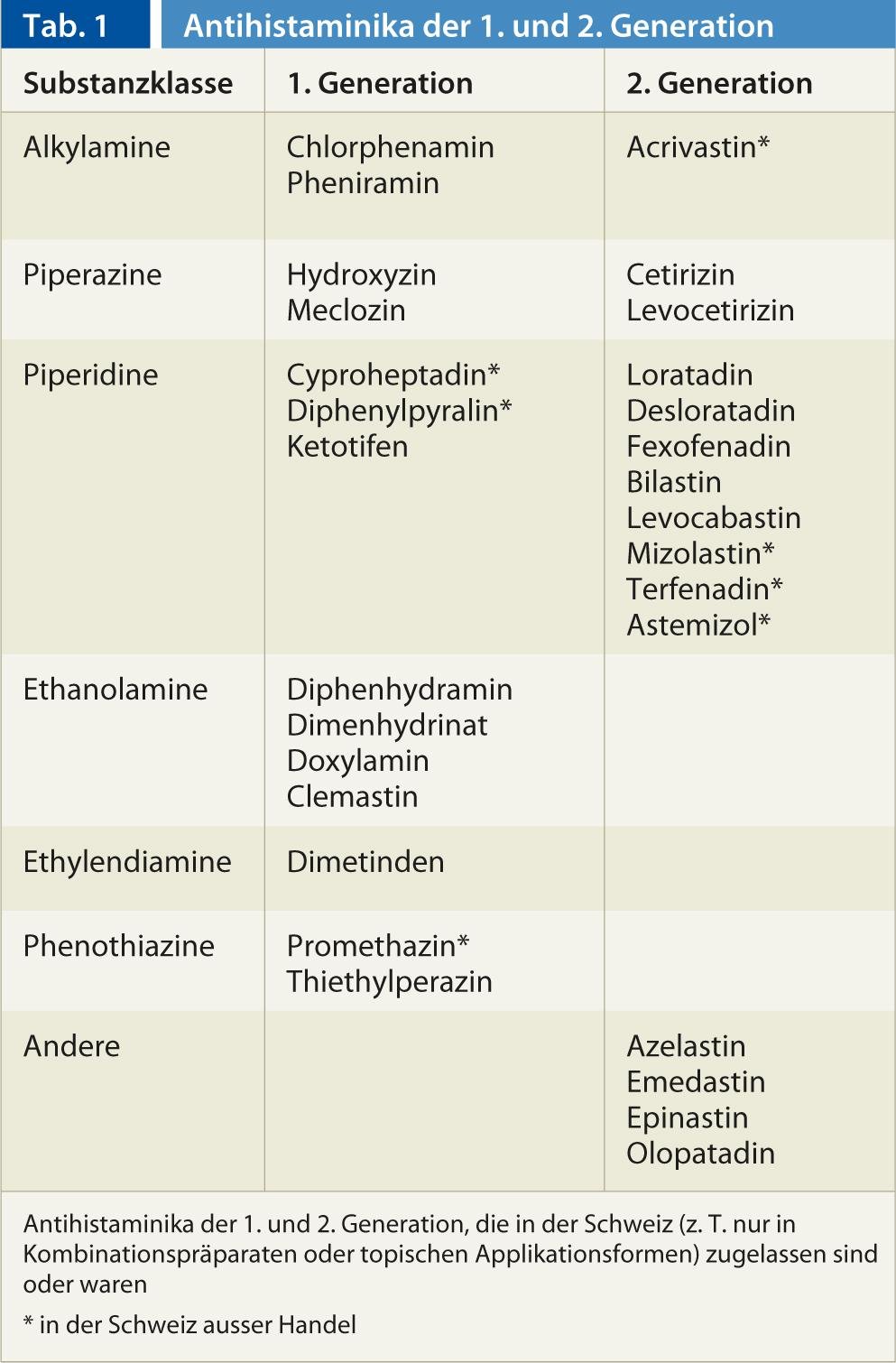
Complessivamente, l’efficacia clinica degli antistaminici H1 del 1. La generazione H1 è stata poco studiata negli studi clinici, mentre le prove per l’uso degli antistaminici H1 della generazione H1 sono state poco studiate. La seconda generazione è indicata per la rinite allergica, la congiuntivite allergica e l’orticaria. L’uso di prodotti del La seconda generazione nella dermatite atopica, nell’asma, nell’anafilassi, nell’angioedema non allergico, nel raffreddore, nel prurito di origine non allergica, ecc. è stata scarsamente studiata negli studi o gli studi non hanno mostrato effetti convincenti e non esiste nemmeno un’approvazione per tali indicazioni. Nei bambini, le sostanze del La prima generazione può portare a effetti collaterali talvolta minacciosi, per cui l’indicazione deve essere fatta con particolare attenzione. In una cosiddetta terza generazione, gli enantiomeri o i metaboliti delle molecole del di seconda generazione, senza che vi siano differenze farmacodinamiche importanti, in modo che le sostanze siano farmacologicamente simili alle sostanze di seconda generazione. Seconda generazione.
Farmacocinetica degli antistaminici H1
Il primo ostacolo che un farmaco deve superare per essere efficace è l’assunzione. Garantire la conformità o l’aderenza è quindi di particolare importanza nella pratica medica quotidiana. La maggior parte degli antistaminici H1 per uso sistemico sono disponibili come forme di dosaggio solide (compresse, dragées, compresse rivestite con film, supposte) o gocce per la somministrazione orale. Alcune sostanze vengono applicate localmente (ad esempio, le gocce per gli occhi). Solo alcuni possono essere somministrati anche per via endovenosa (dimetinden, clemastina, tietilperazina). Il riassorbimento, che è rapido per la maggior parte degli antistaminici H1 di seconda generazione e porta a livelli di picco dopo una o tre ore, è seguito dalla distribuzione nel sangue e nei tessuti, dal metabolismo, se necessario, e dall’escrezione.
Esistono differenze importanti tra gli antistaminici, soprattutto nella metabolizzazione e nell’eliminazione (Tab. 2). Inoltre, ci sono differenze nel metabolismo tra le persone causate da influenze genetiche e ambientali. In particolare, l’enzima CYP2D6 del citocromo P450, coinvolto nella degradazione di alcuni antistaminici H1 (Tab. 2) , mostra una forte variabilità genetica, che può andare dall’assenza alla moltiplicazione della normale attività enzimatica. Inoltre, il CYP2D6 può essere inibito da alcune sostanze: il bupropione, il cinacalcet, gli inibitori selettivi della ricaptazione della serotonina, la paroxetina, la fluoxetina e la duloxetina, nonché il farmaco antimicotico terbinafina sono tra gli inibitori più forti del CYP2D6. Tuttavia, poiché tutti gli antistaminici H1 sono degradati da più di un enzima e spesso vengono eliminati per via renale invariati, la variabilità genetica e l’inibizione del CYP2D6 giocano molto raramente un ruolo nell’uso degli antistaminici H1.
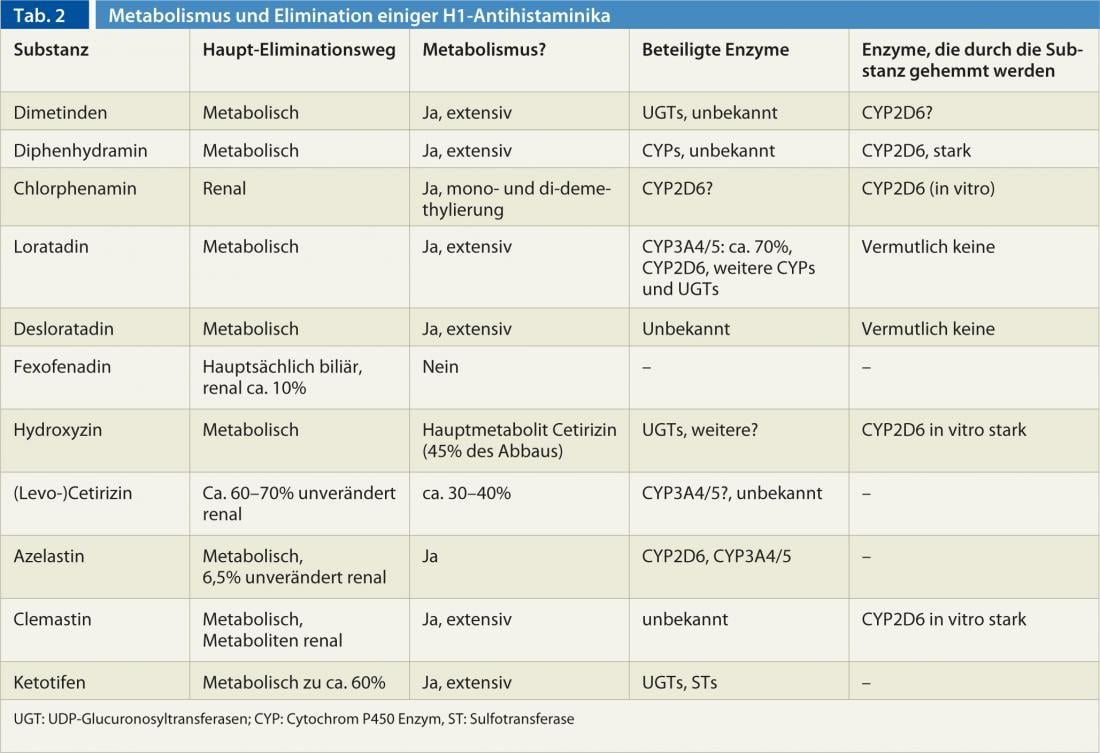
Alcuni antistaminici H1 di prima generazione sono anche inibitori del CYP2D6. (Tab. 2), per cui quando i substrati del CYP2D6 (ad esempio codeina, destrometorfano, molti antipsicotici [aloperidolo, risperidone, aripiprazolo], atomoxetina, molti antidepressivi [la maggior parte dei triciclici, venlafaxina, ecc.], così come i beta-bloccanti metoprololo, carvedilolo e timololo) vengono utilizzati in concomitanza, si deve scegliere una dose inferiore del substrato per evitare effetti avversi.
Nel complesso, la farmacocinetica delle sostanze della prima generazione di è conosciuta solo in modo incompleto. La generazione è conosciuta solo in modo incompleto, poiché le sostanze sono state approvate decenni fa con una quantità di dati notevolmente inferiore a quella necessaria oggi. Ma ci sono anche delle lacune nella conoscenza dei preparati di seconda generazione: La desloratadina viene metabolizzata nel metabolita altrettanto attivo 3-idrossi-desloratadina, ma l’enzima o gli enzimi coinvolti sono sconosciuti, anche se è stato riscontrato che il 2% degli europei e fino al 20% degli africani non possono formare la 3-idrossi-desloratadina. Gli enzimi CYP2D6 e CYP3A4/5 coinvolti nel metabolismo della loratadina non sembrano essere responsabili di questo.
Il CYP3A4 è l’enzima del citocromo P450 più importante, sia in termini di quantità che di numero di substrati. Sebbene non siano note varianti genetiche che alterano la funzione del CYP3A4 (nonostante le intense ricerche), esistono farmaci che aumentano l’attività dell’enzima (rifampicina, efavirenz, fenitoina, carbamazepina, ingredienti dell’iperico, ecc.) e la inibiscono (antifungini azolici, eritromicina, claritromicina, ritonavir, verapamil, diltiazem, amiodarone, ecc.)(antifungini azolici, eritromicina, claritromicina, ritonavir, verapamil, diltiazem, amiodarone, ingredienti del pompelmo, soprattutto nel succo di pompelmo, ecc.) Il CYP3A5, invece, che essenzialmente scompone gli stessi farmaci del CYP3A4, non è quasi mai presente negli europei a causa di una variante genetica, mentre è solitamente funzionale negli africani. Ad eccezione della loratadina, dell’azelastina e probabilmente anche della cetirizina, il CYP3A4/5 non svolge un ruolo negli antistaminici.
È stato ancora più sorprendente quando è stato dimostrato un effetto del succo di pompelmo in uno studio con la fexofenadina: Quando la fexofenadina è stata assunta insieme al succo di pompelmo, i livelli di fexofenadina sono diminuiti, soprattutto poco dopo l’assunzione, rispetto all’assunzione con acqua (ci si sarebbe aspettato un aumento se il CYP3A4 fosse stato inibito dalle furanocumarine del succo di pompelmo). Questo effetto può essere spiegato dal fatto che altre sostanze presenti nel succo di pompelmo (il flavonoide naringina) inibiscono il trasportatore intestinale OATP1A2, necessario per l’assorbimento della fexofenadina dal lume intestinale.
Un’altra interazione che non è stata ancora completamente chiarita riguarda anche la fexofenadina: la somministrazione contemporanea di itraconazolo ha portato a livelli di fexofenadina molte volte superiori . Poiché la fexofenadina non viene metabolizzata dal CYP3A, che è potentemente inibito dall’itraconazolo, l’interazione è stata spiegata con l’inibizione della P-glicoproteina, il trasportatore che si ritiene svolga un ruolo importante nell’eliminazione della fexofenadina. Se questa ipotesi sia corretta e se anche altri antimicotici azolici portino ad aumenti dei livelli, deve ancora essere indagato.
D’altra parte, va notato che sostanze come la (levo-)cetirizina vengono eliminate principalmente per via renale, per cui le limitazioni della funzione renale devono portare anche a riduzioni della dose. Per la levocetirizina, per esempio, è necessario ridurre la dose a partire da una tasso di filtrazione glomerulare inferiore a 50 mL/min, si prescrive una somministrazione di 5 mg ogni 2 giorni; in caso di compromissione della funzione renale più grave, l’intervallo di dosaggio deve essere ulteriormente prolungato.
In Svizzera, le informazioni relative al paziente sugli effetti collaterali e le interazioni, gli aggiustamenti della dose e altri problemi legati ai farmaci possono essere ottenute dalle strutture di farmacologia clinica degli ospedali universitari.
Effetti avversi degli antistaminici: Focus tempo QTc
Mentre la maggior parte degli effetti avversi degli antistaminici H1 sono dovuti alla loro azione sul recettore H1 (affaticamento, riduzione delle prestazioni cognitive e psicomotorie, aumento dell’appetito) o (nel caso delle sostanze più vecchie) dagli effetti sul recettore della m-acetilcolina (secchezza delle fauci, ritenzione urinaria, tachicardia), sul recettore alfa-adreno (ipotensione, vertigini, tachicardia riflessa) o sul recettore della serotonina (ad esempio, aumento dell’appetito). Mentre gli effetti degli antistaminici H1 possono essere spiegati dai loro effetti sul recettore alfa-adrenergico (ipotensione, vertigini, battito cardiaco riflesso) o sul recettore della serotonina (ad esempio, aumento dell’appetito), è meno noto che alcuni antistaminici H1 inibiscono anche i canali ionici cardiaci, in particolare il canale IKr, che modula il rapido deflusso del potassio durante la ripolarizzazione e può quindi portare a un prolungamento della ripolarizzazione (dell’intervallo QT nell’ECG) e persino alla torsades de pointes, la tachicardia ventricolare.
Nel complesso, il potenziale degli antistaminici H1 di prolungare il tempo QTc nell’ECG è poco studiato. L’interesse di per questo effetto collaterale potenzialmente fatale è stato suscitato quando ci sono state diverse segnalazioni di episodi di torsades de pointes per i primi antistaminici H1 di seconda generazione, terfenadina e astemizolo. Nella maggior parte dei casi , sono state assunte dosi eccessive o non sono state osservate interazioni che aumentano la concentrazione. Le due sostanze sono state ritirate dal mercato nel 1990 a causa di aritmie ventricolari. Per la loratadina, la fexofenadina e la cetirizina, sono stati riportati casi individuali di prolungamento del tempo QTc e in alcuni casi di tachicardia torsades de pointes.
I fattori di rischio generali per il prolungamento del tempo QTc sono illustrati nella Tabella 3 . Sembra quindi consigliabile, soprattutto nei pazienti a rischio a cui vengono somministrati antistaminici regolarmente e a dosi elevate, effettuare un ECG e controllare il potassio e il magnesio nel siero.

CONCLUSIONE PER LA PRATICA
- Gli antistaminici H1 di seconda generazione sono generalmente ben tollerati.
- Il prolungamento del tempo QTc è possibile, soprattutto in caso di sovradosaggio e di fattori di rischio.
- Il succo di pompelmo non deve essere bevuto con la fexofenadina, poiché riduce le concentrazioni di fexofenadina. L’itraconazolo aumenta le concentrazioni di fexofenadina, il che può portare a sintomi di sovradosaggio.
- Nel caso di antistaminici escreti principalmente a livello renale, come la (levo-)cetirizina, la dose deve essere adattata in caso di insufficienza renale.
PD Alexander Jetter, MD
Letteratura:
- Simons FE, Simons KJ: Istamina e H1-antistaminici: un secolo di progressi. J Allergy Clin Immunol 2011; 128: 1139-1150.
- Shon JH, Yoon YR, Hong WS, Nguyen PM, Lee SS, Choi YG, Cha IJ, Shin JG: Effetto dell’itraconazolo sulla farmacocinetica e farmacodinamica della fexofenadina in relazione al polimorfismo genetico MDR1. Clin Pharmacol Ther 2005; 78: 191-201.
- Banfield C, Gupta S, Marino M, Lim J, Affrime M: Il succo di pompelmo riduce la biodisponibilità orale della fexofenadina ma non della desloratadina. Clin Pharmacokinet 2002; 41: 311-318.
- Hondeghem LM, Dujardin K, Hoffmann P, Dumotier B, De Clerck F: Il prolungamento del QTc indotto da un farmaco sottostima pericolosamente il potenziale proaritmico: lezioni dalla terfenadina. J Cardiovasc Pharmacol 2011; 57: 589-597.
- Compalati E, Baena-Cagnani R, Penagos M, Badellino H, Braido F, Gómez RM, Canonica GW, Baena-Cagnani CE: Revisione sistematica sull’efficacia della fexofenadina nella rinite allergica stagionale: una meta-analisi di studi clinici randomizzati, in doppio cieco, controllati con placebo. Int Arch Allergy Immunol 2011; 156: 1-15.
PRATICA GP 2013; 8(3): 14-17