L’interessamento polmonare nel contesto di una malattia reumatica infiammatoria sistemica è una manifestazione relativamente frequente, che richiede una conoscenza adeguata sia da parte degli pneumologi che dei reumatologi e idealmente una cura interdisciplinare di questi casi. Si possono trovare diverse patologie polmonari parenchimatose, per cui le alterazioni granulomatose devono essere intese come un sottogruppo tra queste.
L’interessamento polmonare nel contesto di una malattia reumatica infiammatoria sistemica è una manifestazione relativamente frequente, che richiede una conoscenza adeguata sia da parte degli pneumologi che dei reumatologi e idealmente una cura interdisciplinare di questi casi. Si possono trovare diverse patologie polmonari parenchimatose, per cui le alterazioni granulomatose devono essere intese come un sottogruppo tra queste. Il granuloma si riferisce ad un accumulo nodulare circoscritto di cellule infiammatorie nel tessuto, classicamente da macrofagi, ma anche da granulociti o linfociti. Quando si riscontra un’evidenza istologica di malattia polmonare granulomatosa, è necessario elaborare diverse diagnosi differenziali reumatologiche. L’obiettivo di questo articolo è quello di fornire una panoramica delle malattie polmonari granulomatose in ambito reumatologico, ad eccezione della sarcoidosi. L’attenzione è rivolta alla diagnostica e, soprattutto, alla terapia.
Vasculiti associate all’ANCA
Le vasculiti associate agli ANCA sono le prime da considerare. Questo gruppo di vasculiti dei piccoli vasi comprende tre entità: Granulomatosi con poliangioite (GPA), poliangioite microscopica (MPA) e granulomatosi eosinofila con poliangioite (EGPA), senza formazione di granuloma dal punto di vista istologico nella MPA.
Granulomatosi con poliangioite (GPA), quadro clinico
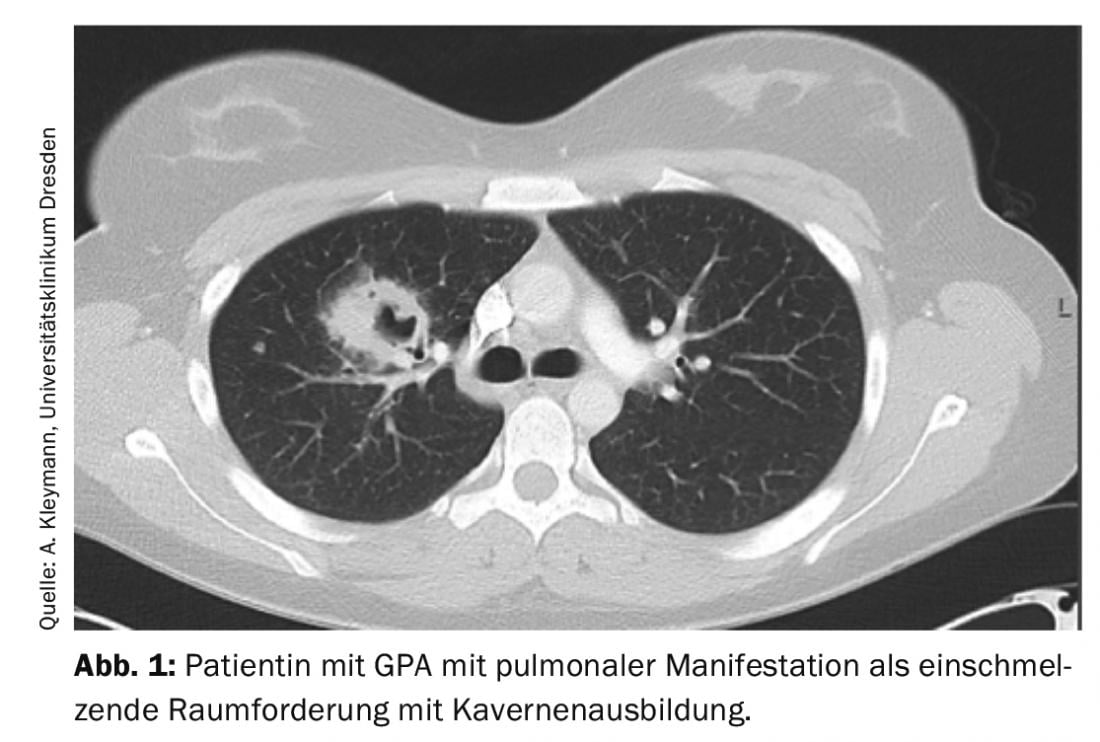
La GPA può essere inizialmente oligosintomatica (stadio localizzato della malattia). Il coinvolgimento del rinofaringe nel senso di una sinusite o rinite cronica è tipico all’inizio. Con l’interessamento polmonare, la GPA è già classificata come malattia generalizzata, per cui ci si devono aspettare sintomi clinici di accompagnamento (sintomi B, mialgie, artralgie, rash cutanei, artrite) e sintomi paraclinici (elevazione della CRP e della VES, anemia, leucocitosi, trombocitosi). Il coinvolgimento polmonare è possibile sotto forma di noduli polmonari, infiltrati, fibrosi polmonare, caverne o addirittura emorragia alveolare (Fig. 1).
Diagnosi ANCA
Una parte importante della diagnosi è la determinazione degli ANCA (anticorpi antineutrofili citoplasmatici) con autoanticorpi diretti contro la proteinasi-3 e la mieloperossidasi. Gli ANCA sono stati descritti per la prima volta nel 1982 nella glomerulonefrite e inizialmente sono stati valutati come associati a un virus. Solo nel 1985 è stata descritta nei pazienti con GPA. Inizialmente, l’immunofluorescenza indiretta è raccomandata come gold standard. I risultati devono essere classificati in base al modello di fluorescenza e al titolo. pANCA è un modello di fluorescenza perinucleare, cANCA è un modello citoplasmatico (Fig. 2). A volte il pattern di fluorescenza viene riportato come xANCA, ma si presume che si tratti di ANCA non specifici, ad esempio nel contesto della colite ulcerosa.
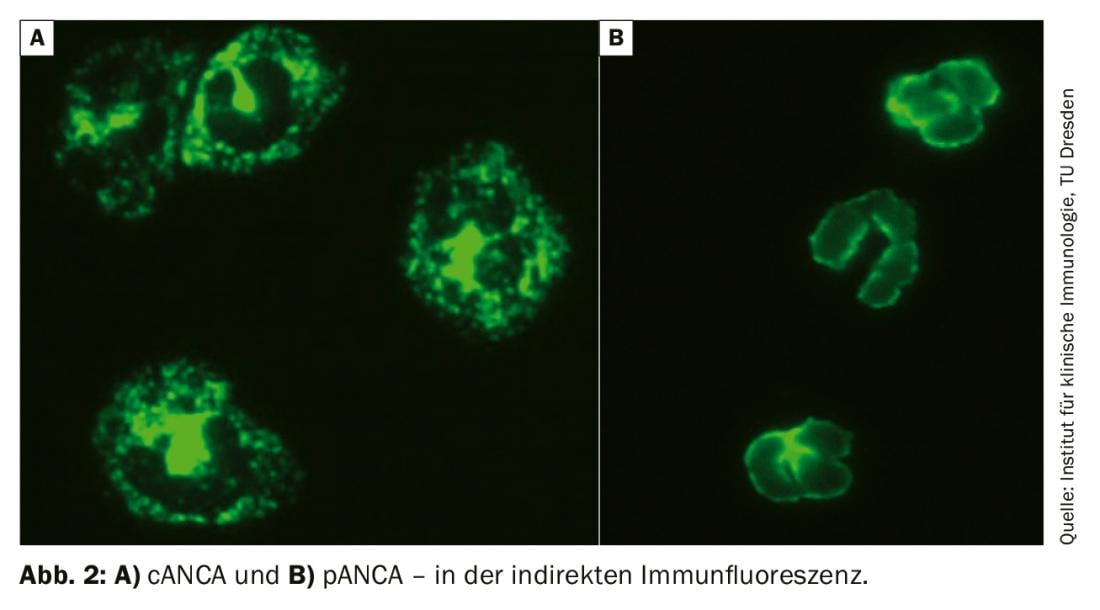
La specificità dei risultati ANCA positivi è aumentata solo dalla rilevazione di autoanticorpi contro gli antigeni target corrispondenti, nel caso di pANCA contro la mieloperossidasi e nel caso di cANCA contro la proteinasi-3. La loro determinazione viene solitamente effettuata tramite ELISA (enzyme-linked immunosorbent assay). Se si sospetta una vasculite associata agli ANCA, è necessario avviare uno screening strutturato per altre possibili manifestazioni d’organo. In particolare, si deve considerare il coinvolgimento renale sotto forma di glomerulonefrite immune di Pauci. Pertanto, oltre ai parametri di ritenzione (creatinina, volume urinario, GFR), deve essere richiesto senza eccezioni un sedimento urinario. In caso di eritrocituria, proteinuria o evidenza di cilindri ialini, si deve ordinare la microscopia delle urine con la domanda di eritrociti dismorfici nel senso di un sedimento nefritico attivo e la quantificazione delle proteine. Se si sospetta una manifestazione renale, è necessaria una biopsia renale. Questo spesso non solo conferma la diagnosi, ma determina anche la prognosi della funzione renale. Nei pazienti obesi o a maggior rischio di sanguinamento, la biopsia renale transgiugulare è un’alternativa ragionevole alla biopsia transcutanea.
Terapia
Dopo la diagnosi, nel caso di una manifestazione polmonare di vasculite, la terapia di induzione con ciclofosfamide o rituximab è solitamente indicata in concomitanza con i corticosteroidi, per cui i corticosteroidi devono essere ridotti in tempi relativamente brevi, in modo che la dose sia di 7,5-10 mg/d prednisolone equivalente dopo 3 mesi. L’eccezione è rappresentata da una manifestazione isolata con noduli polmonari piccoli, non disgreganti e senza limitazioni funzionali, dove la terapia di base con metotrexato, a condizione che non vi sia insufficienza renale, o azatioprina è possibile anche senza una precedente terapia di induzione.
In base ai dati dello studio CYCLOPS, l’induzione di ciclofosfamide come somministrazione in bolo i.v. è da preferire rispetto alla somministrazione orale, grazie ai bassi tassi di effetti collaterali [1]. Il dosaggio standard è di 15 mg/kg di peso corporeo; le prime 3 dosi vengono somministrate ogni 2 settimane, poi ogni 3 settimane. La dose deve essere regolata in base all’età e alla funzione renale (riduzione a 12,5 mg/kgKG nei 60-70enni se la creatinina arriva a 300 µmol/l, a 10 mg/kgKG nella creatinina >300 µmol/l e nei >70enni se la creatinina arriva a 300 µmol/l, e a 7,5 mg/kgKG nei >70enni se la creatinina >300 µmol/l). Di solito sono sufficienti 6 boli di . In caso di terapia con ciclofosfamide, oltre alla tossicità gonadica e della vescica urinaria (per cui si raccomanda anche la somministrazione di 2-mercaptoethanesulfonatsodium (MESNA)), occorre sottolineare il rischio di leucopenia rilevante. Il nadir si verifica 10 -12 giorni dopo la somministrazione, pertanto è necessario effettuare un controllo emocromocitometrico appropriato in questo periodo e, se necessario, un ulteriore aggiustamento della dose per la somministrazione successiva (se i leucociti <4 GPT/l riduzione della dose di ciclofosfamide del 25% e la somministrazione successiva solo quando la conta leucocitaria >4 GPT/l). Un’alternativa alla ciclofosfamide è l’induzione con l’anticorpo anti-CD20 rituximab. L’efficacia di rituximab è stata dimostrata in 2 studi (RAVE e RITUXVAS). Qui sia il regime con somministrazione 4× settimanale di 375 mg/m2 di superficie corporea che 2× 1 g di dose assoluta ad intervalli di 14 giorni sono una pratica comune. Prima di iniziare la terapia con rituximab, è necessario effettuare uno screening dell’epatite (rischio di riattivazione dell’epatite cronica B) e, idealmente, aggiornare lo stato di vaccinazione, poiché si può ipotizzare una risposta vaccinale ridotta/insufficiente durante la terapia.
L’aumento del rischio di infezione da patogeni atipici non deve essere sottovalutato con entrambi i farmaci, per cui la profilassi dello Pneumocystis jirovecii è necessaria per tutti i pazienti durante la terapia di induzione, indipendentemente dalla sostanza scelta (ad esempio Cotrim [Trimethoprim/Sulfamethoxazol] 480 mg 1× al giorno). La profilassi può essere interrotta dopo il completamento dell’induzione e alla dose di prednisolone <10-15 mg/d. Il ruolo della plasmaferesi nei decorsi gravi come l’emorragia alveolare diffusa (DAH) continua ad essere considerato controverso. La Società Europea di Reumatologia (EULAR) raccomanda di prenderla in considerazione nella sua ultima raccomandazione del 2016; tuttavia, lo studio PEXIVAS pubblicato sul NEJM nel febbraio 2020, che ha incluso 31 pazienti con DAH grave nel braccio di scambio di plasma e 30 pazienti nel braccio di studio senza plasmaferesi, non ha mostrato alcun beneficio in termini di sopravvivenza [2].
Dopo il completamento dell’induzione, la terapia di mantenimento deve essere solitamente effettuata con rituximab (2 dosi di 500 mg per via endovenosa a intervalli di 14 giorni, seguite da 500 mg ogni 6 mesi) o in alternativa con azatioprina (2 mg/kg di peso corporeo) per almeno 2 anni. Quando si inizia la terapia con azatioprina, inizialmente sono necessari regolari controlli di laboratorio (emocromo e transaminasi) e la co-somministrazione di inibitori della xantina ossidasi (allopurinolo e febuxostat) è controindicata. Inoltre, in alcuni centri si raccomanda la determinazione della tiopurina S-metiltransferasi (TPMT) per evitare una grave soppressione del midollo osseo nei pazienti con la corrispondente carenza enzimatica.
Le alternative in caso di controindicazione/intolleranza sono il metotrexato (ma non in caso di coinvolgimento renale con insufficienza renale) o il micofenolato mofetile. Tuttavia, va sottolineato che il micofenolato mofetile non è approvato per questo scopo ed è quindi un “uso off-label” e anche meno efficace dell’azatioprina in base ai dati dello studio IMPROVE (hazard ratio per la ricaduta con micofenolato mofetile vs. azatioprina 1,69) [3]. Un nuovo e interessante approccio terapeutico è il blocco del recettore del complemento C5a con l’avacopan [4]. Nello studio presentato online al Congresso Europeo sul Reumatismo EULAR di quest’anno, questa sostanza somministrata per via orale ha mostrato un effetto paragonabile a quello del gruppo di controllo con steroidi, quando è stata utilizzata in concomitanza con la terapia standard con ciclofosfamide o rituximab, ma senza steroidi. Pertanto, in futuro, si potrebbe rinunciare completamente agli steroidi in questo gruppo di pazienti, utilizzando Avacopan.
Granulomatosi eosinofila con poliangioite (EGPA)
L’EGPA è impegnativa nel gruppo delle vasculiti associate agli ANCA. È difficile confermare la diagnosi perché l’istologia spesso non rileva la vasculite, gli ANCA sono spesso negativi e si deve escludere una sindrome ipereosinofila primaria come espressione di una malattia clonale.
Una tipica manifestazione polmonare è la comparsa di infiltrati volatili. Istologicamente, l’eosinofilia tissutale può essere spesso rilevata nella biopsia polmonare. Anche la decisione sulla terapia non è sempre facile a causa di studi insufficienti. In assenza di parametri prognostici sfavorevoli, come l’interessamento renale o cardiaco, è ipotizzabile una monoterapia con corticosteroidi, altrimenti sono disponibili la ciclofosfamide per le manifestazioni che minacciano gli organi o il metotrexato e l’azatioprina per i decorsi più lievi. Il rituximab è di secondaria importanza nell’EGPA a causa dei dati insufficienti. D’altra parte, è interessante l’uso dell’antagonista dell’IL-5 mepolizumab, che ha mostrato efficacia in uno studio di fase 3 su 136 pazienti con EGPA refrattaria o recidivante.
Un maggior numero di pazienti è stato in grado di raggiungere la remissione con la somministrazione s.c. di mepolizumab per 4 settimane. 300 mg di mepolizumab per 52 settimane, più pazienti hanno raggiunto la remissione (32% contro 3%) e hanno anche ridotto la dose di prednisolone al di sotto di 4 mg/d (44% contro 7%). Tuttavia, anche nel gruppo mepolizumab, la remissione non è stata raggiunta nel 47% dei casi [5]. Il mepolizumab è ora approvato dalla FDA anche per l’EGPA.
Noduli reumatici
I noduli reumatoidi mostrano un’altra formazione di granuloma. È più probabile che si verifichino nei pazienti con artrite reumatoide sieropositiva di lunga data (fattore reumatoide e/o anticorpi anti-CCP positivi) e noduli reumatoidi cutanei. La differenziazione dalla malignità è difficile senza l’istologia, poiché da un lato ci possono essere diversi noduli reumatoidi che progrediscono di dimensioni nel corso della malattia, e dall’altro i pazienti con artrite reumatoide per se hanno un rischio maggiore di malignità. In genere, i noduli reumatici polmonari sono localizzati a livello subpleurico o nell’area dei setti interlobulari e di solito sono asintomatici (Fig. 3).
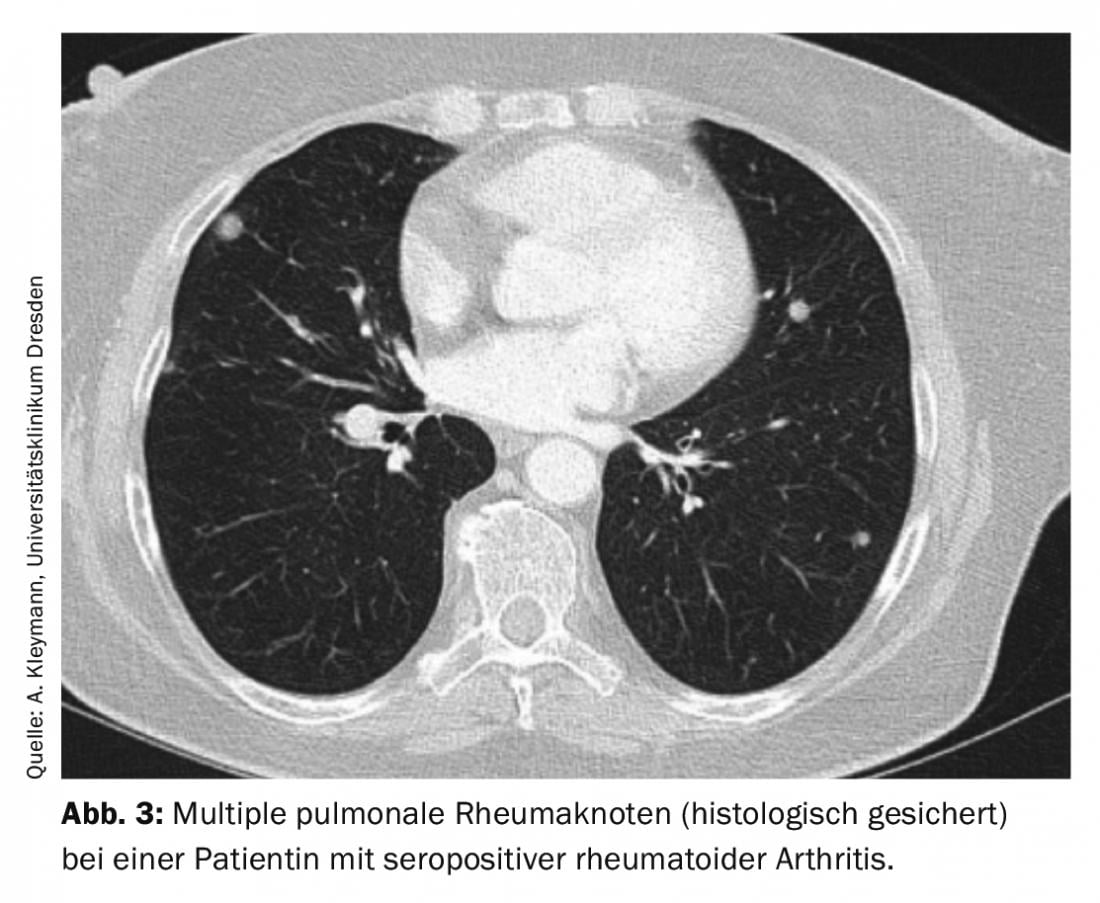
Tuttavia, possono anche causare versamento pleurico, pneumotorace, emottisi o addirittura fistole broncopolmonari. Se vengono rilevati noduli reumatoidi, potrebbe essere necessario modificare la terapia di base, in quanto con il metotrexato si osserva spesso un aumento delle dimensioni dei noduli reumatoidi. Con la leflunomide sono stati descritti anche casi di sviluppo di noduli reumatoidi polmonari [6,7].
Infezioni atipiche
Nel caso di una reazione infiammatoria granulomatosa, si devono considerare anche le infezioni atipiche sotto la terapia di base più immunosoppressiva. Dal punto di vista infettivologico, le infezioni fungine come l’istoplasmosi e la sporotricosi devono essere considerate in aggiunta alla tubercolosi. Il rischio di infezione nei pazienti con malattie reumatiche infiammatorie sistemiche è aumentato sia dalla malattia di base che dai terapici di base immunosoppressivi utilizzati.
Soprattutto, i bloccanti del TNF sembrano avere un’influenza rilevante sul rischio di infezioni polmonari granulomatose. Ciò è ben compreso dall’importanza fisiopatologica del TNF-alfa nella difesa e nella formazione del granuloma contro gli agenti patogeni batterici e fungini.
Istoplasmosi
L’istoplasmosi polmonare è causata da Histoplasma capsulatum. Questo patogeno ubiquitario si trova principalmente in Nord e Centro America. La differenziazione clinica da sarcoidosi e malignità è spesso difficile, perché le manifestazioni polmonari sono diverse (infiltrati polmonari o focolai rotondi, caverne, linfoadenopatia medistinale o lesioni che occupano spazio) (Fig. 4).
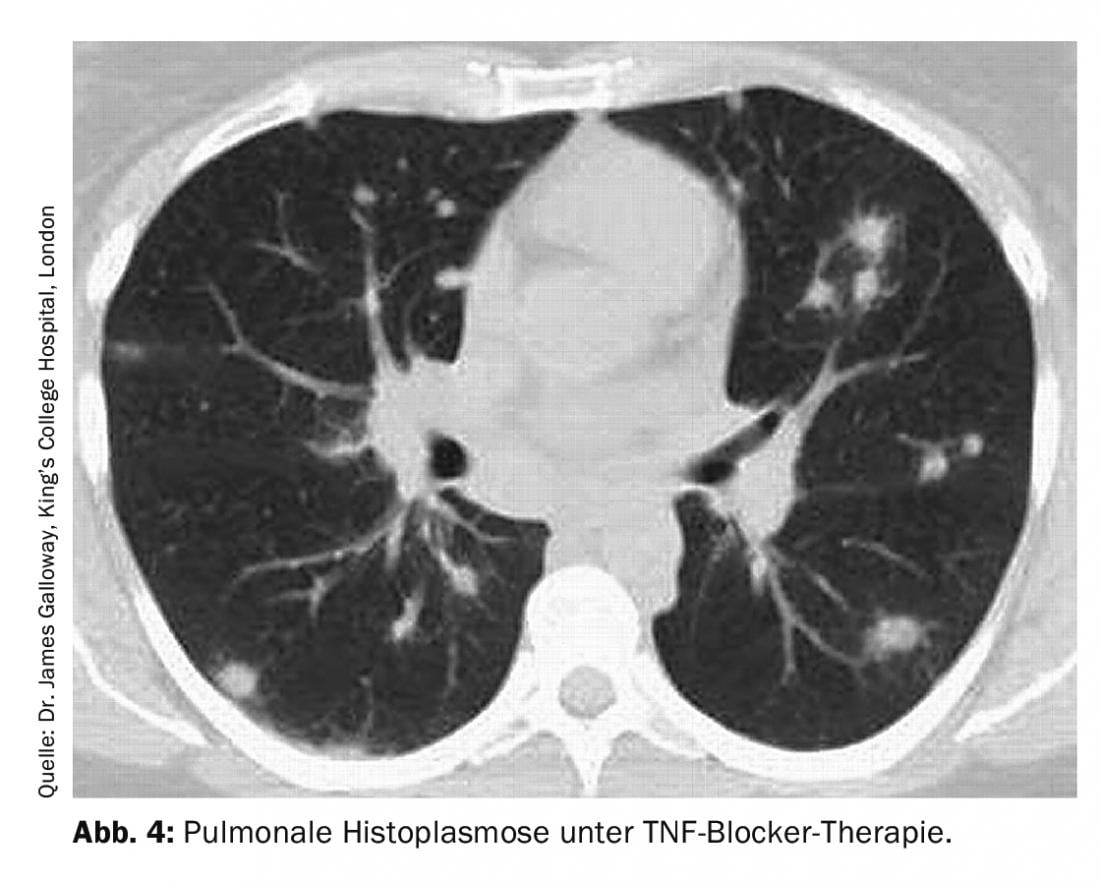
Oltre alla sierologia (anticorpi specifici per l’istoplasma e determinazione dell’antigene), vengono utilizzati a livello diagnostico il lavaggio bronco-alveolare con colorazione fungina e la coltura, ma anche il corrispondente esame istologico del bioptato per i funghi. Dal punto di vista terapeutico, non si utilizza alcuna terapia o vari antimicotici, a seconda della gravità della malattia.
Sporotricosi
La sporotricosi è causata da Sporothrix schenckii e ha classicamente una manifestazione cutanea. Nelle manifestazioni polmonari, la malattia assomiglia alla tubercolosi (Fig. 5) . Il gold standard della diagnosi è la rilevazione culturale dei patogeni, poiché l’istopatologia può essere falsamente negativa con un basso numero di patogeni anche con una colorazione appropriata, e i test sierologici e di PCR non sono ancora disponibili di routine.
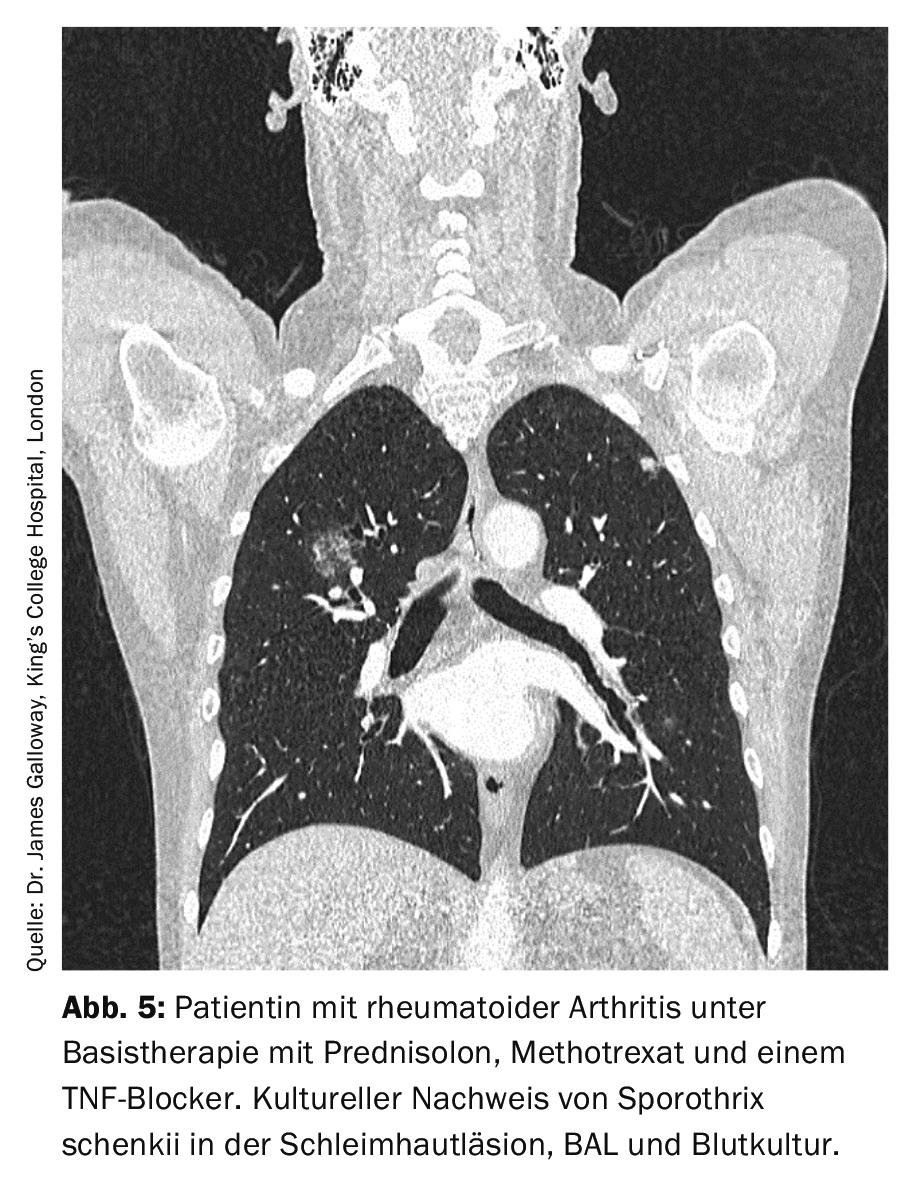
Se non trattata, la sporotricosi polmonare è fatale, quindi è sempre necessaria una terapia antimicotica. Nei corsi lievi, si può usare il p.o.. Può essere utilizzato l’itraconazolo 2×200 mg/d. Nel caso di una manifestazione grave, si raccomanda prima di tutto l’amfotericina B per via endovenosa e il passaggio all’itraconazolo avviene solo in corso d’opera. La durata della terapia deve essere di almeno 1 anno [8].
Immunodeficienze primarie
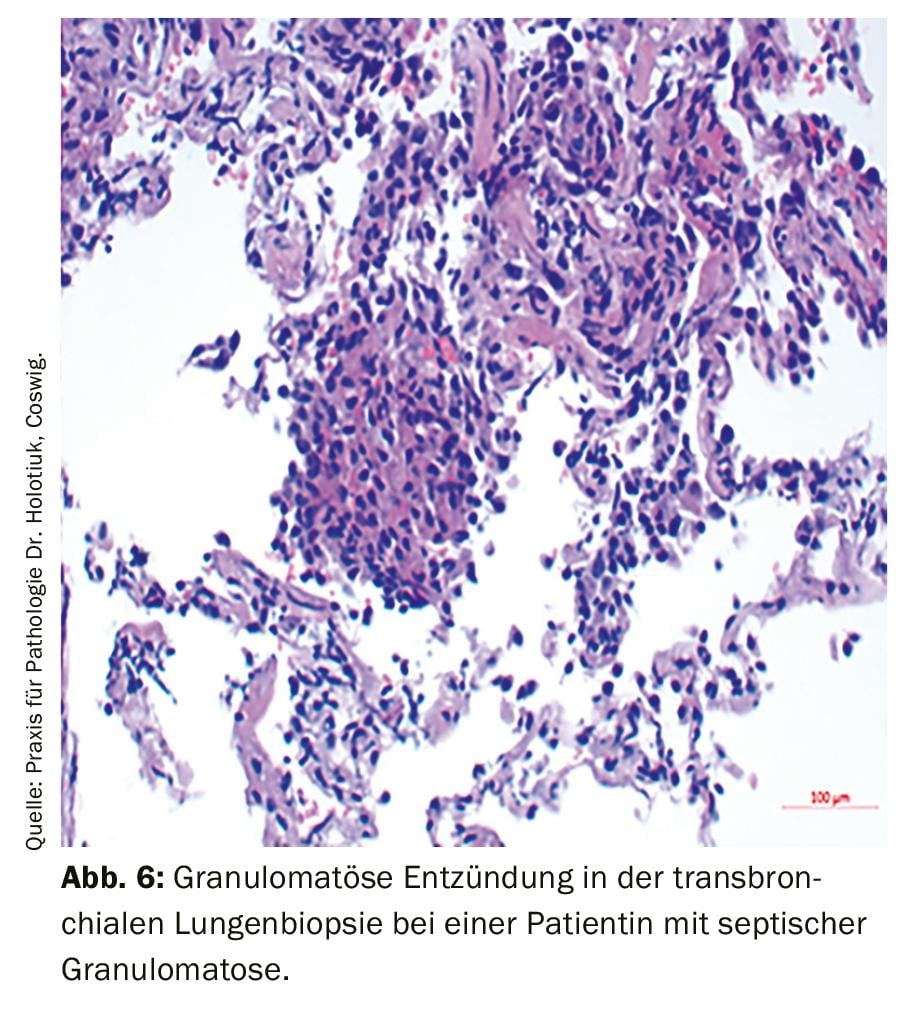
Infine, va menzionato il complesso gruppo delle immunodeficienze primarie. In particolare, i pazienti con infezioni ricorrenti e fenomeni autoimmuni devono essere presi in considerazione per questa diagnosi differenziale. L’acronimo ELVIS descrive la suscettibilità patologica alle infezioni (agenti patogeni e localizzazioni atipiche, decorso prolungato, intensità e numero di infezioni eccezionali [Summe]). L’acronimo GARFIELD riassume le possibili manifestazioni come espressione di un disturbo della regolazione immunitaria: Granulomi (Fig. 6), fenomeni autoimmuni, febbre ricorrente, alterazioni cutanee eczematose, linfoproliferazione (linfoadenopatia, splenomegalia) e infiammazione intestinale cronica [9]. Se c’è un sospetto giustificato, devono essere avviate ulteriori diagnosi con uno stato immunitario cellulare (emocromo differenziale, tipizzazione dei linfociti) e umorale (determinazione delle immunoglobuline, se necessario anche con le sottoclassi IgG, fattori del complemento) e deve essere avviato un chiarimento appropriato da parte di un medico esperto nella diagnostica e nel trattamento delle immunodeficienze.
Messaggi da portare a casa
- In caso di alterazioni polmonari granulomatose, si devono considerare anche le malattie reumatologiche come la vasculite dei piccoli vasi, i noduli reumatoidi, ma anche le infezioni atipiche sottoposte a terapia di base e le immunodeficienze primarie con fenomeni autoimmuni.
- Se si sospetta una vasculite polmonare, si deve iniziare la diagnostica ANCA con la determinazione degli anticorpi contro la proteinasi 3 e la mieloperossidasi e, se la diagnosi è confermata, si deve iniziare subito un’immunosoppressione adeguata.
- I noduli reumatoidi polmonari non sono rari nell’artrite reumatoide sieropositiva, ma spesso sono difficili da distinguere dai tumori maligni quando si manifestano per la prima volta.
- Le infezioni polmonari atipiche devono essere prese in considerazione soprattutto nei pazienti trattati con biologici e/o con steroidi ad alto dosaggio.
- Le immunodeficienze primarie si verificano frequentemente con i fenomeni autoimmuni e devono quindi essere escluse se c’è un sospetto fondato.
Letteratura:
- de Groot K, Harper L, Jayne DR, et al: Ciclofosfamide orale ad impulsi rispetto a quella giornaliera per l’induzione della remissione nella vasculite associata agli anticorpi anticitoplasma antineutrofili: uno studio randomizzato. Ann Intern Med 2009; 150(10): 670-680.
- Walsh M, et al: Scambio di plasma e glucocorticoidi nella vasculite grave associata ad ANCA. N Engl J Med 2020; 382: 622-631.
- Hiemstra TF, Walsh M, Mahr A, et al: Micofenolato mofetile vs azatioprina per il mantenimento della remissione nella vasculite associata agli anticorpi anticitoplasma antineutrofili: uno studio randomizzato controllato. JAMA 2010; 304(21): 2381-2388.
- Merkel PA, Jayne DR, Wang C, et al: Valutazione della sicurezza e dell’efficacia di Avacopan, un inibitore del recettore C5a, nei pazienti con vasculite associata ad anticorpi antineutrofili trattati in concomitanza con rituximab o ciclofosfamide/azatioprina: protocollo per uno studio di fase 3, randomizzato, in doppio cieco, controllato dall’attività. JMIR Res Protoc 2020; 9(4): e16664.
- Wechsler ME, Akuthota P, Jayne D, et al: Mepolizumab o placebo per la granulomatosi eosinofila con poliangioite. N Engl J Med 2017; 376(20): 1921-1932.
- Horvath IF, Szanto A, Csiki Z, et al: Noduli reumatoidi intrapolmonari in un paziente con artrite reumatoide di lunga data trattato con leflunomide. Pathol Oncol Res 2008; 14(1): 101-104.
- Rozin A, Yigla M, Guralnik L, et al: Nodulosi polmonare reumatoide e osteopatia associata alla terapia con leflunomide. Clin Rheumatol 2006; 25(3): 384-388.
- Kauffman CA, Bustamante B, Chapman SW, et al: Linee guida di pratica clinica per la gestione della sporotricosi: aggiornamento 2007 della Infectious Diseases Society of America. Clin Infect Dis 2007; 45(10): 1255-1265.
- Linea guida AWMF “Diagnosticare la presenza di un’immunodeficienza primaria” Stato: 31.10.2017, valido fino al 31.10.2020.
InFo PNEUMOLOGIA & ALLERGOLOGIA 2020; 2(3): 12-16