Un gonfiore periodico, molto doloroso ed esteso dei linfonodi può anche indicare una rara malattia di Castleman. La forma multicentrica è una malattia sistemica grave che non è facile da diagnosticare.
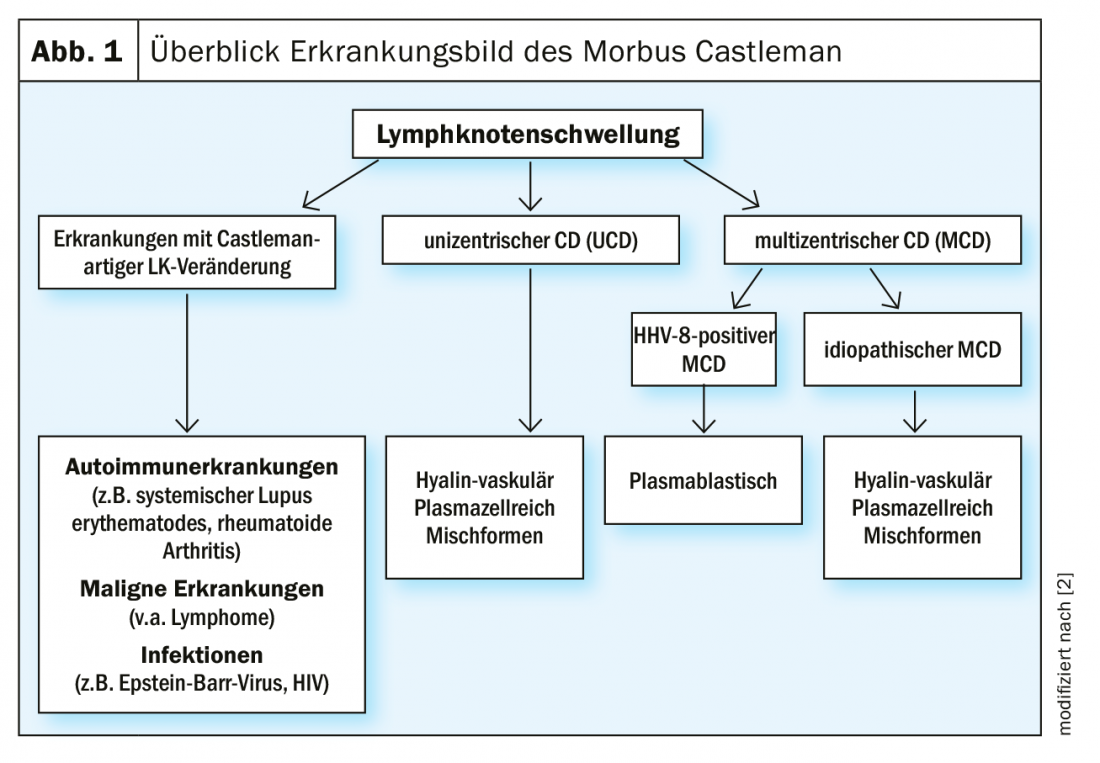
In caso di gonfiore esteso dei linfonodi che si verifica periodicamente, molto doloroso e distinto, si deve pensare anche alla malattia di Castleman. Questa malattia rara si divide in malattia di Castleman unicentrica (UCD) e forme multicentriche e spesso viene trascurata. L’UCD è una forma benigna della malattia con iperplasia localizzata del tessuto linfatico [1]. In questo caso, l’escissione del linfonodo interessato è il metodo di scelta [2]. Tuttavia, diventa più problematico con la malattia di Castleman multicentrica (MCD). Si tratta di una grave malattia sistemica con una prognosi spesso sfavorevole. Una sovrapproduzione di diverse citochine, in particolare dell’interleuchina-6 (IL-6), determina una sintomatologia eterogenea. Esiste una stretta associazione con il virus dell’herpes umano 8 (HHV-8). Questo, a sua volta, è in gran parte congruente con le infezioni da HIV. Di conseguenza, oltre il 90% dei pazienti infettati dall’HIV sono anche positivi all’HHV-8. Al contrario, nei pazienti HIV-negativi, il tasso di è solo del 10-15% – in questo caso predomina la MCD idiopatica [3] (Fig. 1).
Epidemiologia
Fondamentalmente, tutte le forme di malattia di Castleman si verificano molto raramente, e una grande percentuale non viene certamente mai diagnosticata correttamente. Si stima che ci siano 2,4 casi per milione di abitanti, che corrispondono a circa 20 casi in Svizzera [4]. Con il 35,4%, la MCD sembra essere meno frequente della UCD (64,6%) [5]. Tuttavia, l’incidenza è più alta nei pazienti con infezione da HIV. Secondo uno studio britannico, si tratta di 4,3 casi per 10.000 anni-paziente, con una leggera tendenza all’aumento negli ultimi anni [2,6]. Negli studi osservazionali, la mortalità nei primi 5 anni dopo la diagnosi è stata del 30-35% [7]. Secondo una revisione sistematica, la sopravvivenza libera da malattia nell’iMCD dopo 3 anni era del 45,7% [8].
Patogenesi
La forma multicentrica con ingrossamento di numerosi linfonodi è stata descritta per la prima volta nel 1978. Si verifica più frequentemente nei pazienti sieropositivi. L’HHV8 è responsabile della malattia di Castleman multicentrica in quasi tutti i pazienti HIV-positivi e di circa la metà delle malattie nei pazienti HIV-negativi [9]. Producono un’interleuchina virale che è molto simile all’IL-6 umana e ha effetti simili. Deve solo legarsi a una delle due subunità del recettore dell’IL-6 per essere attiva. Questo spiega l’effetto significativamente più ampio sulle cellule bersaglio e probabilmente anche le tipiche “tempeste di citochine” [10,11,2]. L’interleuchina-6 e altre citochine proinfiammatorie inducono la proliferazione delle cellule B e delle plasmacellule, la secrezione di fattori di crescita endoteliale vascolare e l’angiogenesi.
Le cause dell’iMCD, invece, sono ancora oggetto di un dibattito controverso. Gli scienziati ipotizzano che una malattia infiammatoria autoimmune, un’infezione virale e una predisposizione genetica possano essere responsabili [12]. Oltre all’IL-6, sono coinvolte altre molecole di segnalazione, tra cui VEGF (“fattore di crescita dell’endotelio vascolare”), TNF-alfa e interleuchina-1 [13]. La proliferazione linfocitaria nell’iMCD è di solito policlonale e una conseguenza dell’iper-citochinemia [14,2]. Tuttavia, la proliferazione monoclonale è stata osservata in alcune lesioni del sottotipo vascolare ialino [15,2].
Sintomi e disturbi
I sintomi si presentano in modo molto eterogeneo, il che spesso rende difficile una diagnosi efficace. Tuttavia, la malattia è generalmente accompagnata da dolore nei linfonodi colpiti. Questi gonfiori si verificano spesso in episodi, soprattutto nella MCD HHV-8 positiva. Inoltre, ci sono chiari sintomi B come febbre, sudorazione notturna e perdita di peso. Quasi tutti i pazienti lamentano debolezza e sensazione di malessere, nausea, vomito e perdita di appetito. Anche la milza e il fegato sono ingrossati. Sono presenti anche epatomegalia, sintomi respiratori e una tendenza all’edema con ipalbuminemia. Di solito c’è una grave anemia ematologica, che spesso può essere accompagnata da una massiccia trombocitopenia.
In genere, la malattia progredisce in episodi che durano da pochi giorni a settimane, durante i quali i pazienti hanno spesso febbre alta e sono gravemente malati. In mezzo, ci sono periodi più lunghi che possono durare settimane, durante i quali i pazienti si sentono di nuovo molto meglio. Le dimensioni dei linfonodi possono quasi tornare alla normalità. La frequenza e l’intensità delle ricadute in genere aumentano nel tempo, ma sono noti anche decorsi autolimitanti. Tuttavia, esiste un rischio significativamente aumentato di linfomi maligni, soprattutto di sottotipi di linfoma piuttosto rari, come i linfomi plasmablastici o i linfomi da effusione primaria [2,12].
L’iMCD si manifesta con un sintomo di affaticamento pronunciato, soprattutto nelle fasi iniziali. A differenza della MCD, i sintomi come febbre, gonfiore dei linfonodi e anemia sono molto meno pronunciati e il decorso delle ricadute è meno brusco. I sintomi possono variare da lievi sintomi costituzionali a decorsi pericolosi per la vita, con anasarca e insufficienza multipla degli organi. Un sottotipo di iMCD grave, la sindrome di TAFRO, è stato definito di recente. Si tratta di un complesso sintomatologico di trombocitopenia, ascite, febbre, fibrosi reticolare nel midollo osseo e organomegalia con γ-globulina normale . Inoltre, sono comuni le associazioni con la sindrome POEMS, un quadro clinico composto da neuropatia periferica, organomegalia, endocrinopatia, gammopatia monoclonale e alterazioni cutanee [2].
Diagnostica
La malattia di Castleman deve essere differenziata dai linfomi e da altre malattie gravi. La forma grave, in particolare, viene spesso scambiata per un linfoma. L’esame istopatologico di un linfonodo estirpato è obbligatorio. Una puntura di solito non è sufficiente. Il preparato mostra una linfoadenite reattiva con proliferazione delle plasmacellule nella polpa e cambiamenti regressivi nei centri germinali, che di solito sono stratificati come bucce di cipolla e intervallati da vasi. Nella MCD HHV-8 positiva, di solito si vede un’immagine plasmablastica, spesso con un cosiddetto motivo a tarme nell’area della zona del mantello. Il caratteristico motivo a buccia di cipolla è spesso parzialmente distrutto. L’HHV-8 viene rilevato tramite la colorazione di LANA-1 (“latency-associated-nuclear-antigen”). Il sangue prelevato durante un episodio di malattia mostra livelli elevati di IL-6 e CRP. La triade di sintomi B, viremia HHV-8 e reperti istologici è utile per la diagnosi [17].
La diagnosi di iMCD è molto più difficile, in quanto devono essere escluse molte altre malattie. Le alterazioni istopatologiche si riscontrano anche, ad esempio, in diverse infezioni (compreso il virus di Epstein-Barr) o nel lupus eritematoso, nell’artrite reumatoide, nella sindrome di Sjögren e in alcune neoplasie, in particolare i linfomi maligni. Pertanto, oltre ai cambiamenti istopatologici, si devono sempre prendere in considerazione i parametri clinici e di laboratorio. L’iMCD si differenzia in tre sottotipi istopatologici: il tipo ialino-vascolare, il tipo plasmacellulare e il tipo misto. Tutti si verificano in circa il 20-40% dei casi [18]. Il tipo ialino-vascolare è caratterizzato da cellule dendritiche follicolari (FDC) displastiche e centri germinali atrofici, intervallati da vasi ialini, attorno ai quali si dispongono concentricamente i linfociti. Nel tipo plasmacellulare, i centri germinali sono iperplastici piuttosto che atrofici e le FDC e l’architettura linfonodale sono normali [2].
Terapia
La malattia di Castleman unicentrica ha una prognosi molto buona, in quanto il linfonodo colpito può essere rimosso chirurgicamente. La recidiva della malattia è estremamente rara. Tuttavia, poiché nella MCD sono colpiti molti linfonodi, l’intervento chirurgico non è efficace. Il trattamento rimane difficile. Non esiste una terapia standard per i pazienti con malattia di Castleman multicentrica. Concettualmente, vengono utilizzate sostanze immunosoppressive e citotossiche. I glucocorticoidi sono efficaci, ma spesso non portano a un sollievo dei sintomi a lungo termine (Fig. 2).
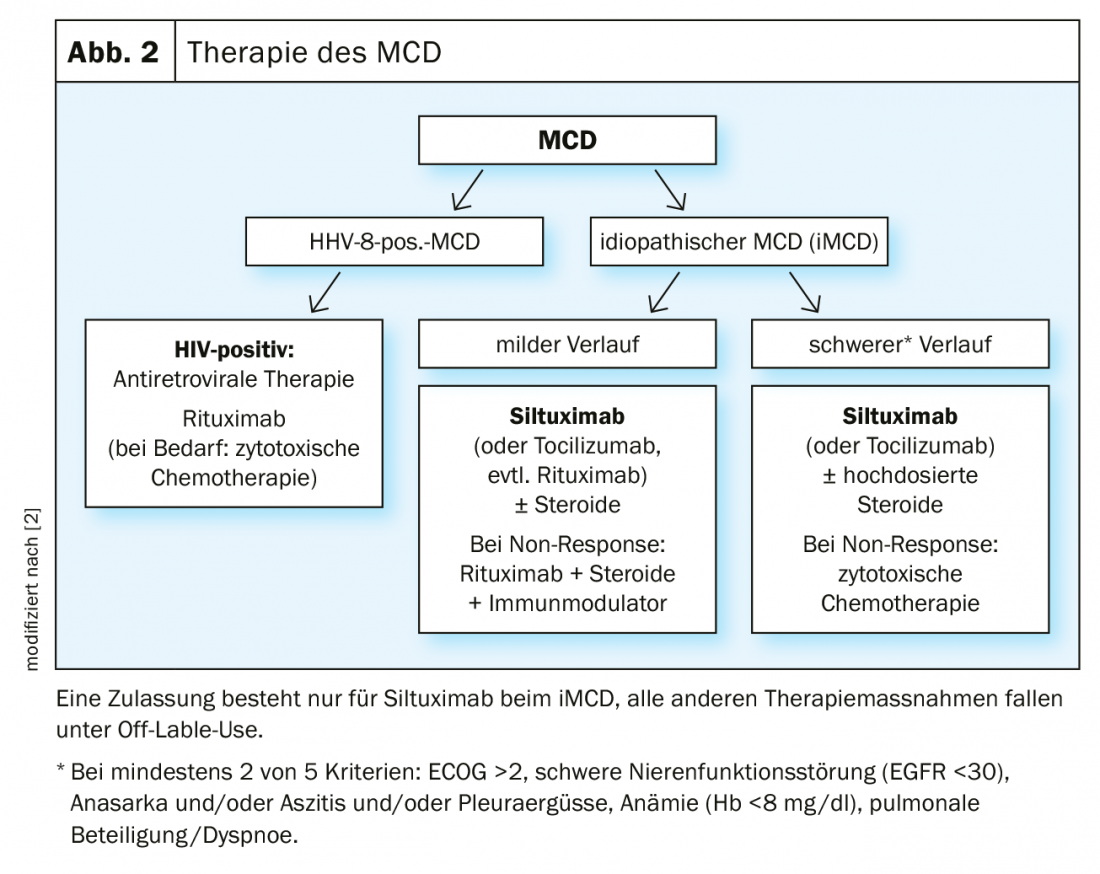
Nell’iMCD, si utilizza principalmente il blocco della via di segnalazione dell’IL-6 mediante anticorpi monoclonali come il siltuximab. Nella MCD HHV-8 positiva, l’attenzione si concentra sull’eliminazione citotossica delle cellule responsabili della sovrapproduzione di citochine. Le terapie immunomodulanti sono principalmente gli steroidi, oltre a sostanze antivirali come il valganciclovir. Dal 2018, per la prima volta esistono raccomandazioni basate sul consenso per l’iMCD [19]. Si basano sull’esperienza di 344 pazienti sottoposti a un totale di 479 terapie e tengono conto della gravità della malattia, delle terapie precedenti e della risposta.
L’unico farmaco approvato finora per il trattamento dell’iMCD è il siltuximab, un anticorpo monoclonale contro l’IL-6 umana. Viene utilizzato per tutte le forme di iMCD con o senza steroidi. Nello studio pivotal, 79 pazienti con iMCD sono stati trattati con siltuximab o placebo per tre settimane [20,7]. L’endpoint primario era una risposta duratura in termini di dimensioni del tumore e di miglioramento del punteggio dei sintomi clinici per almeno 18 settimane. Mentre non è stata osservata alcuna risposta nel gruppo placebo, i tassi nel gruppo verum sono stati del 34%. Siltuximab è stato complessivamente ben tollerato. Gli effetti collaterali caratteristici e frequenti sono prurito cutaneo, infezioni del tratto respiratorio superiore, esantema ed edema locale, prevalentemente di gravità 1 e 2. Gli effetti collaterali gravi sono affaticamento (12%) e sudorazione notturna (8%). Tuttavia, il tasso in questo caso non era superiore a quello del placebo, per cui è più probabile che questi sintomi siano attribuiti alla malattia di base [7].
Il tocilizumab, un anticorpo contro il recettore dell’IL-6, è considerato un’alternativa al siltuximab nelle raccomandazioni internazionali, ma è approvato solo in Europa per l’artrite reumatoide.
Anche il rituximab, un anticorpo monoclonale contro il CD20, non è approvato per il trattamento della MCD. Soprattutto i pazienti con MCD HHV-8 positivo sembrano rispondere bene. In diverse serie di casi, è stato osservato un miglioramento significativo della sopravvivenza globalea e della sopravvivenza libera da malattia. Le citochine, ma anche la CRP, le immunoglobuline e la carica virale HHV-8 sono diminuite [2,21]. Tuttavia, in circa un terzo dei casi, si è verificata la progressione del sarcoma di Kaposi, che spesso accompagna la malattia. In questo caso, potrebbe essere utile una combinazione con sostanze citostatiche con attività KS [22]. In situazioni di pericolo di vita, si deve prendere in considerazione la splenectomia. Riduce il pool di cellule infettate da HHV-8 [2].
Messaggi da portare a casa
- La malattia di Castleman si divide in forma benigna unicentrica (UCD) e malattia di Castleman multicentrica (MCD).
- La MCD è una malattia sistemica che spesso non viene riconosciuta a causa della sua eterogeneità e della sua rarità.
- Il gonfiore doloroso dei linfonodi si verifica in episodi, spesso accompagnati da sintomi B come febbre o sudorazione notturna.
- Siltuximab è attualmente l’unico farmaco approvato per il trattamento della MCD idiopatica.
Letteratura:
- Castleman B, Towne VW: Cartelle cliniche del Massachusetts General Hospital; esercizi clinicopatologici settimanali. N Engl J Med 1954; 251: 396-400
- Hoffmann C, Tiemann M: Malattia di Castleman multicentrica: raramente diagnosticata correttamente. Dtsch Arztebl 2019; 116(46): [32]; DOI: 10.3238/PersOnko.2019.11.15.06
- Hoffmann C: Malattia di Castleman multicentrica (MCD) – un quadro clinico raro, spesso non riconosciuto. Trillium 2015. www.trillium.de/zeitschriften/trillium-krebsmedizin/archiv/ausgaben-2015/heft-12015/multizentrischer-morbus-castleman-mcd-ein-seltenes-oft-verkanntes-krankheitsbild.html (ultimo accesso 06.04.2020)
- Robinson D Jr, Reynolds M, Casper C, et al: Epidemiologia clinica e modelli di trattamento dei pazienti con malattia di Castleman multicentrica: risultati di due centri di trattamento statunitensi. Br J Haematol 2014; 165: 39-48.
- Haap M, Wiefels J, Horger M, et al: Risultati clinici, di laboratorio e di imaging nella malattia di Castleman – Il sottotipo decide. Blood Rev 2018; 32: 225-234.
- Powles T, Stebbing J, Bazeos A, et al.: Il ruolo della soppressione immunitaria e dell’HHV-8 nella crescente incidenza della malattia di Castleman multicentrica associata all’HIV. Ann Oncol 2009; 20: 775-779.
- www.dgho.de/publikationen/stellungnahmen/fruehe-nutzenbewertung/siltuximab/siltuximab-dgho-stellungnahme-20141006.pdf (ultimo accesso 06.04.2020)
- Talat N, Schulte KM: Malattia di Castleman: analisi sistematica di 416 pazienti della letteratura. Oncologo 2011; 16:1316-24
- Fajgenbaum DC, van Rhee F, Nabel CS: Malattia di Castleman multicentrica idiopatica HHV-8-negativa: nuove intuizioni su biologia, patogenesi e terapia. Sangue 2014; 123: 2924-2933. DOI: 10.1182/blood-2013-12-545087.
- Li H, Wang H, Nicholas J. Rilevamento del legame diretto dell’interleuchina-6 codificata dall’herpesvirus umano 8 (vIL-6) sia con la gp130 che con il recettore dell’IL-6 (IL-6R) e identificazione dei residui aminoacidici della vIL-6 importanti per la segnalazione dipendente e indipendente dall’IL-6R. J Virol 2001; 75: 3325-3334.
- Moore PS, Boshoff C, Weiss RA, Chang Y: Mimetismo molecolare dei geni delle citochine umane e del percorso di risposta alle citochine da parte del KSHV. Science 1996; 274: 1739-1744.
- https://medlexi.de/Morbus_Castleman (ultimo accesso 06.04.2020)
- Pierson S, Stonestrom A, Ruth J, et al.: La quantificazione delle proteine plasmatiche di riacutizzazioni e remissioni della malattia di Castleman multicentrica idiopatica rivela una ‘tempesta di chemochine’ e separa i sottotipi clinici (abstract). Sangue 2017; 130(Suppl. 1).Abstract 3592
- Ohyashiki JH, Ohyashiki K, Kawakubo K, et al. Analisi genetica molecolare, citogenetica e immunofenotipica Malattia di Castleman di tipo plasmacellulare. Am J Clin Pathol 1994; 101: 290-295.
- Chang KC, Wang XC, Hung YL: Monoclonalità e anomalie citogenetiche nella malattia di Castleman vascolare ialina. Mod Path 2014; 7: 823-831.
- Iwaki N, Fagenbaum D, Nabel CS, et al: L’analisi clinicopatologica della sindrome TAFRO dimostra un sottotipo distinto di malattia di Castleman multicentrica HHV-8-negativa. Am J Hematol 2016; 91: 220-226.
- Bower M, Pria AD, Coyle C, et al: Schemi di criteri diagnostici per la malattia di Castleman multicentrica in 75 casi. J Acquir Immune Defic Syndr 2014; 65(2): e80-82.
- Liu AY, Nabel CS, Finkelman BS, et al: Malattia di Castleman multicentrica idiopatica: una revisione sistematica della letteratura. Lancet Hematol 2016; 3: e163-175.
- van Rhee F, Voorhees P, Dispenzieri A, et al: Linee guida di consenso internazionali, basate sull’evidenza, per il trattamento della malattia di Castleman multicentrica idiopatica. Sangue 2018; 132: 2115-2124.
- van Rhee F, Wong RS, Munshi N, et al: Siltuximab per la malattia di Castleman multicentrica: uno studio randomizzato, in doppio cieco, controllato con placebo. Lancet Oncol 2014; 15: 966-974, DOI: 10.1016/S1470-2045(14)70319-5.
- Bower M, Veraitch O, Szydlo R, et al: Cambiamenti delle citochine durante la terapia con rituximab nella malattia di Castleman multicentrica associata all’HIV. Sangue 2009; 113: 4521-4524.
- Uldrick TS, Polizzotto MN, Aleman K, et al: Rituximab più doxorubicina liposomiale in pazienti infetti da HIV con malattia di Castleman multicentrica associata a KSHV. Sangue 2014; 124: 3544-3552.
InFo ONCOLOGIA & EMATOLOGIA 2020; 8(2): 4-7