L’atrofia muscolare spinale è una rara malattia genetica caratterizzata dalla perdita di motoneuroni nel midollo spinale e nella parte inferiore del tronco encefalico. Nel frattempo, gli esperti concordano sul fatto che la gestione della terapia deve essere orientata alle esigenze individuali e agli obiettivi soggettivi della persona interessata, soprattutto con l’avanzare dell’età. Perché questi hanno una grande influenza sulla qualità della vita.
Un neonato su circa 10.000 ha un difetto genetico che interrompe la trasmissione degli impulsi dei motoneuroni. L’atrofia muscolare spinale (SMA) è una malattia muscolare rara, prevalentemente autosomica recessiva, in cui le cellule del corno anteriore del motore e i nuclei dei nervi cranici motori degenerano. È la malattia ereditaria più comune che provoca la morte in età infantile e spesso viene diagnosticata in età molto precoce – anche se non esclusivamente [1,2]. La SMA può anche manifestarsi per la prima volta in età adulta [3]. Questa eterogeneità richiede quindi una terapia individuale che tenga conto dell’attività della malattia e delle esigenze del paziente.
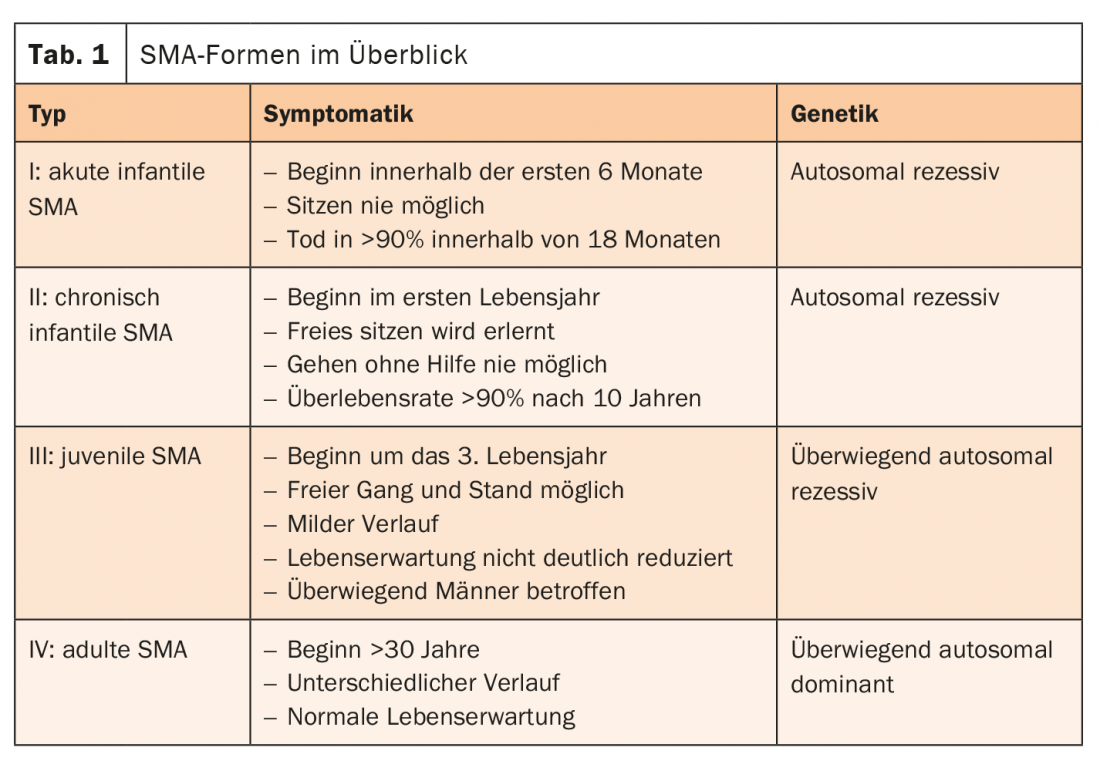
La causa della SMA è solitamente un difetto del gene SMN1. Insieme al gene SMN2, forma la proteina “Survival of Motor Neuron” (SMN). Questo gioca un ruolo centrale nella trasmissione degli impulsi dalle cellule nervose ai muscoli. Se il gene SMN1 viene meno, la proteina importante può essere prodotta solo dal gene SMN2 rimanente. Pertanto, più copie di SMN2 ci sono, più tardivo è l’esordio della SMA e più favorevole il decorso. I pazienti con SMA ad esordio tardivo (tipo II-IV) hanno spesso un’aspettativa di vita normale. Le forme di SMA si distinguono in base al modello di distribuzione, all’insorgenza della malattia, alla gravità e al modello di ereditarietà (Tab. 1) [4].
Eseguire lo screening dei neonati, se possibile
Una diagnosi definitiva di SMA può essere fatta solo attraverso un test genetico. Tuttavia, poiché la SMA di tipo 1, se non trattata, porta alla morte o richiede la ventilazione meccanica permanente nel 90% dei casi entro il compimento dei due anni di età del paziente, è indicata la diagnosi precoce della malattia e l’inizio più rapido possibile del trattamento. La gestione della terapia è complessa e comprende l’assistenza in fase acuta e le misure di riabilitazione, ortopedia, supporto respiratorio, fisioterapia e interventi farmacologici.
Successi terapeutici anche con adolescenti e adulti
Il primo farmaco che non agisce solo a livello sintomatico, ma affronta anche le cause della malattia correggendo il difetto genetico sottostante, è stato approvato nel 2017 con il nusinersen. L’oligonucleotide antisenso (ASO) è un modulatore di splicing specifico che lascia il genoma così com’è e migliora la funzione naturale della proteina SMN2. Questo permette di formare quantità maggiori di proteina SMN completa e funzionale. Ora sono stati presentati i primi dati di uno studio di coorte multicentrico. L’obiettivo principale di questo studio osservazionale non interventistico è quello di indagare gli obiettivi e le aspettative di trattamento dei pazienti adulti affetti da 5q-SMA, nonché la soddisfazione soggettiva dei pazienti per il trattamento [5].
I risultati preliminari mostrano che gli obiettivi di trattamento individuali sono di particolare importanza. All’interno dei diversi tipi di 5q-SMA, gli obiettivi della terapia variano in modo significativo. Pertanto, la conservazione della funzione del braccio è uno degli obiettivi terapeutici più comuni, con una predominanza nei pazienti di tipo 1 e di tipo 2. Nei pazienti con SMA di tipo 3, la conservazione e il miglioramento della funzione delle gambe sono più spesso prioritari. La terapia con ASO sembra essere adatta a questo scopo. I risultati di ulteriori studi mostrano anche che varie abilità motorie possono stabilizzarsi o addirittura sono possibili miglioramenti motori clinicamente rilevanti negli adulti con 5q-SMA [6-8].
Congresso: DGM 2021
Letteratura:
- Bowerman M, et al: Strategie terapeutiche per l’atrofia muscolare spinale: SMN e oltre. Dis Model Mech 2017; 10: 943-954.
- Borasio G, et al: Diagnostica delle atrofie muscolari spinali. Neurologia 2001; 20:113-118.
- www.sma-schweiz.ch/spinale-muskelatrophie/typen-der-proximalen-sma (ultimo accesso 05.04.2021)
- www.muskelgesellschaft.ch/diagnosen/spinale-muskelatrophien-sma (ultimo accesso 05.04.2021)
- Meyer, et al: Congresso DGN 2020, P333.
- Hagenacker T, et al: Lancet Neurol 2020; 19(4): 317-325.
- Walter MC, et al: J Neuromuscul Dis 2019; 6: 453-465.
- Maggi L, et al: JNNP. 2020, Nov;91(11): 1166-1174.
InFo NEUROLOGY & PSYCHIATRY 2021; 19(3): 41 (pubblicato il 5.6.21, in anticipo sulla stampa).
PRATICA GP 2021; 16(8): 45