Cosa bisogna considerare per quanto riguarda la diagnosi e il trattamento degli adenomi ipofisari? In che misura la distinzione tra incidentalomi ormono-secernenti e ormono-inattivi è rilevante per il trattamento? Durante la sessione di endocrinologia dei Medidays di quest’anno, sono state fornite informazioni su aspetti importanti del work-up degli adenomi ipofisari.
Gli incidentalomi sono lesioni trovate per caso durante esami di imaging per altre malattie o sintomi non specifici (ad esempio, mal di testa). La prevalenza degli incidentalomi dell’ipofisi varia a seconda dei dati dello studio (TAC, risonanza magnetica, autopsia) ed è compresa tra il 4 e il 40%, con la maggior parte di dimensioni inferiori a 10 mm, ha detto il dottor Tschopp, MD. La stragrande maggioranza dei tumori ipofisari incidenti sono adenomi, cioè tumori benigni delle cellule parenchimatiche del lobo anteriore dell’ipofisi [1]. Tuttavia, il relatore ha sottolineato che la diagnosi differenziale è molto ampia e che la collaborazione tra endocrinologi e neuroradiologi è importante. L’eziologia comprende altre neoplasie (ad esempio craniofaringomi, germinomi, metastasi o linfomi, molto raramente carcinomi), malattie infiammatorie/granulomatose (neurosarciodosi, istiocitosi, ascesso, ipofisite) e alterazioni cistiche (cisti di Rathke, cisti colloide).

Linee guida cliniche
Secondo le linee guida cliniche, oltre all’imaging sellare mirato, il seguente work-up dovrebbe essere eseguito in tutti i pazienti con un incidentaloma dell’ipofisi (compresi quelli senza sintomi) [2]:
- Esame clinico e test di laboratorio per l’ipofunzione ormonale (ipopituitarismo)
- Esame clinico e di laboratorio per l’iperfunzione ormonale (ipersecrezione ormonale).
La diagnosi deve basarsi sulle seguenti tre domande guida:
a) C’è un effetto di massa?
(Cefalea, perdita del campo visivo dovuta alla compressione del chiasma ottico o visione doppia?)
b) Ci sono prove di un’insufficienza ipofisaria?
(Insufficienza del lobo anteriore e/o diabete insipido?)
c) Si tratta di un adenoma ormonale?
(Sindrome di Cushing, acromegalia o iperprolattinemia?)
Ci sono prove di un’insufficienza ipofisaria?
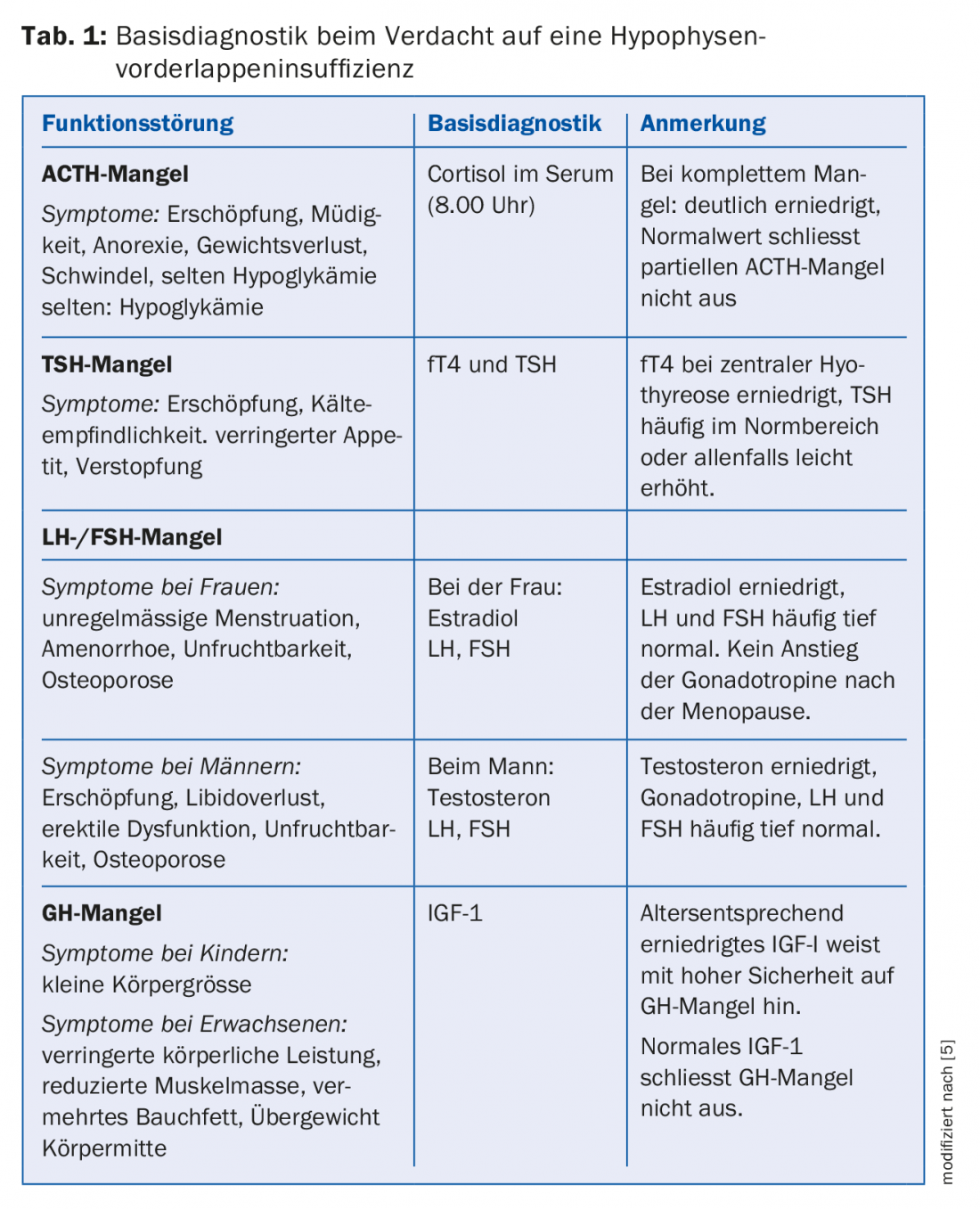
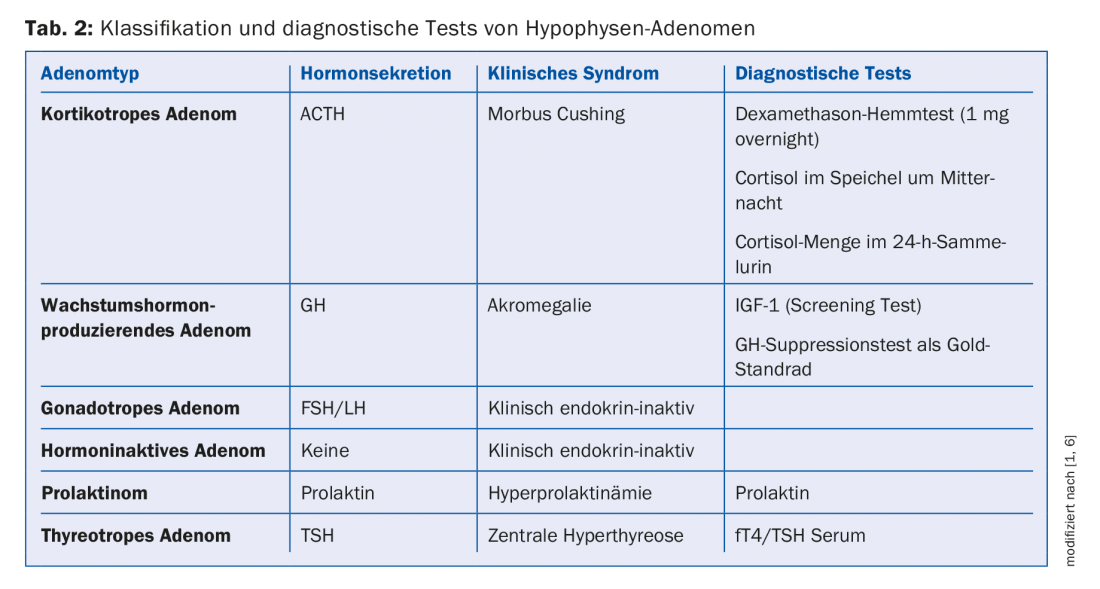
I sintomi dell’insufficienza ipofisaria sono spesso aspecifici (ad esempio, esaurimento, prestazioni ridotte, ecc.) e talvolta si manifestano in modo insidioso nel corso di mesi o anni (Tab. 1). Gli adenomi ipofisari sono la causa più frequente di insufficienza ipofisaria, anche se questo vale soprattutto per i macroadenomi [5]. Se si sospetta un’insufficienza ipofisaria, si devono misurare gli ormoni periferici (cortisolo, fT4, testosterone/estradiolo, IGF1) per un’ulteriore diagnosi. La determinazione esclusiva degli ormoni ipofisari (ACTH, TSH, LH/FSH, GH) non è affidabile, in quanto questi sono spesso nel range di normalità (“inadeguato-normale”) e suggeriscono un asse ormonale intatto, dice il dottor Tschopp, MD. Se si sospetta un’iper- o un’ipofunzione ipofisaria dovuta a un adenoma ipofisario, si possono determinare i seguenti parametri di base: *cortisolo, GH, *IGF-1, TSH, *fT4, LH/FSH, *testosterone/estradiolo e prolattina [1] (*secondo il relatore, gli ormoni più importanti per la diagnosi di un’insufficienza).
I campioni di sangue per la diagnosi devono essere prelevati al mattino presto, poiché il cortisolo e il testosterone in particolare sono soggetti a un forte ritmo circadiano. Se i risultati non sono conclusivi, possono essere necessari ulteriori esami, che di solito vengono eseguiti da uno specialista in endocrinologia [5]. In particolare, è stato sottolineato che la misurazione del cortisolo al mattino è un parametro adeguato nell’insufficienza surrenalica, ma non ha un significato affidabile per quanto riguarda l’iperfunzione (malattia di Cushing); ci sono forti sovrapposizioni di valori negli individui sani e nei pazienti. Si deve anche notare che il diabete insipido con poliuria/dipsia non è un reperto tipico degli adenomi ipofisari, piuttosto questo dovrebbe indirizzare il work-up diagnostico differenziale in un’altra direzione (craniofaringeoma, germinoma, malattie granulomatose).
Si tratta di un adenoma ormonale?
La prolattina e l’IGF-1 sono parametri significativi nella diagnosi di iperfunzione. Se i livelli di prolattina sono normali, si può escludere il prolattinoma. Se i livelli di IGF non sono elevati, l’acromegalia può essere esclusa con un alto grado di probabilità.
Malattia di Cushing: Per la diagnosi della malattia di Cushing sono necessari ulteriori esami: oltre a determinare la quantità di cortisolo nelle urine del prelievo delle 24 ore, si può esaminare il ritmo circadiano misurando il cortisolo nella saliva a mezzanotte o controllare la regolazione dell’autoproduzione di cortisolo (test di soppressione del desametasone, 1 mg durante la notte). Si deve tenere conto che l’aumento dei livelli di cortisolo e i risultati di test patologici possono essere riscontrati nel contesto di altre malattie (ad esempio, pseudo-Cushing nella dipendenza da alcol, depressione, iperglicemia, stress, infezioni). È importante notare che livelli elevati di cortisolo basale non sono una prova della presenza della sindrome di Cushing, e nel caso di livelli normali di cortisolo basale, la sindrome di Cushing non può essere esclusa. Nei pazienti che per la prima volta sono sospettati clinicamente di avere la sindrome di Cushing, devono essere eseguite principalmente le diagnosi di cui sopra e l’imaging iperfisiologico deve essere richiesto solo se l’eccesso è confermato.
Acromegalia: anche l’acromegalia è una malattia rara. Le caratteristiche cliniche dell’acromegalia includono (in modo incompleto): alterazione dei tratti del viso, ingrossamento delle mani e dei piedi, sindrome da apnea notturna, artrite, sindrome del tunnel carpale, iperidrosi, pressione e glicemia elevate. A causa del lento decorso della malattia, l’acromegalia viene spesso diagnosticata solo dopo un periodo di circa dieci anni e, secondo il relatore, può portare a una riduzione significativa dell’aspettativa di vita. Nei pazienti con caratteristiche cliniche tipiche dell’acromegalia, si raccomanda di determinare il livello di IGF-1 come screening. Nei pazienti con incidentaloma, la misurazione dei livelli di IGF-1 è raccomandata anche per escludere in modo affidabile l’acromegalia nei pazienti oligosintomatici. Se c’è una forte evidenza di acromegalia, il sospetto diagnostico può essere confermato da un test di soppressione del GH (test di tolleranza al glucosio orale con 75 g di glucosio).
Prolattinemia: i prolattinomi sono relativamente comuni e rappresentano circa il 30-40% di tutti gli adenomi ipofisari. I prolattinomi sono un caso speciale di adenomi ipofisari, in quanto i prolattinomi possono essere trattati molto bene con i farmaci (agonisti del recettore della dopamina, ad esempio la cabergolina). È stato sottolineato che un livello elevato di prolattina in un incidentaloma non indica automaticamente un prolattinoma; la diagnosi differenziale deve essere considerata l’iperprolattinemia da disinibizione (compressione del peduncolo nel microadenoma). Una causa importante dell’iperprolattinemia è rappresentata dai farmaci e un’anamnesi accurata è essenziale a questo proposito. Livelli elevati di prolattina si riscontrano occasionalmente con i seguenti farmaci: farmaci antipsicotici, inibitori selettivi della ricaptazione della serotonina (SSRI), farmaci usati per trattare la nausea e i disturbi della motilità gastrica. Un’anamnesi farmacologica incompleta. La mancata rivelazione da parte del paziente porta spesso a risultati di iperprolattinemia non chiari e transitori. Si raccomanda di effettuare una determinazione basale della prolattina prima di iniziare una terapia con farmaci antipsicotici, in quanto questo valore può aiutare a restringere la causa nel corso ed evitare chiarimenti inutili.
A causa della complessità della malattia, si raccomanda la cura interdisciplinare dei pazienti da parte di neurochirurghi, neuroradiologi, endocrinologi, oculisti e radioterapisti in un centro con un elevato numero di casi. A questo scopo, l’Ospedale Universitario di Zurigo offre ai colleghi che esercitano la professione privata l’opportunità di presentare i propri casi al Consiglio Interdisciplinare dell’Ipofisi.
Fonte: Medidays, 3-6 settembre 2018, Zurigo
Letteratura:
- Maldaner N, et al: Gestione moderna degli adenomi ipofisari – Stato attuale della diagnosi, del trattamento e del follow-up. Praxis 2018; 107(15): 825-835. doi: 10.1024/1661-8157/a003035.
- Freda PU, et al. Incidentaloma ipofisario: una linea guida di pratica clinica della Società endocrina. J Clin Endocrinol Metab 2011 ; 96(4) : 894-904. doi: 10.1210/jc.2010-1048.
- Molitch ME: Tumori ipofisari non funzionanti e incidentalomi ipofisari. Endocrinol Metab Clin North Am 2008; 37: 151-171.
- Molitch ME: Tumori ipofisari: incidentalomi ipofisari. Best Pract Res Clin Endocrinol Metab 2009; 23: 667-675.
- Möller-Goede DL, Sze L, Schmid C: Ipopituitarismo. Swiss Med Forum 2014; 14(49): 927-931.
- Lake MG, Krook LS, Cruz SV: Adenomi pituitari: una panoramica. Am Fam Physician 2013; 88: 319-327.
PRATICA GP 2018; 13(9): 42-44