
La sindrome di Fanconi-Bickel (FBS) si verifica a causa di varianti nel gene SLC2A2. La diagnosi di una malattia genetica rara può richiedere fino a 5-6 anni, e anche di più nei Paesi a basso e medio reddito con risorse tecnologiche limitate. I medici del Perù hanno presentato il caso di un bambino di due anni e mezzo che non riusciva a crescere, epatomegalia, acidosi metabolica, ipofosfatemia, ipokaliemia e iperlattatemia.
La sindrome di Fanconi-Bickel (OMIM #227810), un disturbo ereditario autosomico recessivo (AR), è caratterizzata da una combinazione di malattie epatiche e renali causate da un difetto nel trasportatore di glucosio GLUT2 (gene SLC2A2). Questo porta all’accumulo di glicogeno, alla disfunzione tubulare renale prossimale e a un’alterata utilizzazione del glucosio e del galattosio.
Il fenotipo comprende mancanza di aumento di peso, distensione addominale, epatomegalia, ipoglicemia a digiuno, iperglicemia postprandiale, glucosuria, fosfaturia, aminoaciduria, poliuria, acidosi metabolica, osteoporosi, ipofosfatemia, rachitismo e presenza di glicogeno nella biopsia epatica o renale. In rari casi, è stato osservato un carcinoma epatocellulare dovuto all’attivazione della via di segnalazione Wnt. Tuttavia, sono stati riportati anche casi di pazienti con sintomi clinici lievi, compresi quelli con glucosuria pura. Il gene SLC2A2 (OMIM *138160) contiene 11 esoni e la sua proteina GLUT2 è composta da 524 aminoacidi e si trova nella membrana cellulare, espressa negli epatociti, negli enterociti, nei tubuli prossimali renali, nelle cellule beta pancreatiche, nei neuroni e negli astrociti. Le varianti patogene di SLC2A2 alterano l’ingresso e l’uscita di glucosio negli epatociti e riducono la secrezione di insulina a causa di una maggiore sensibilità delle cellule beta nella fase postprandiale.
Caso clinico di un bambino di 2 anni e mezzo
Un bambino di 2 anni e 7 mesi, nato e cresciuto in Perù dalla quinta gravidanza di genitori imparentati, si è presentato all’équipe del dottor Hernán Abarca-Barriga dell’Instituto Nacional de Salud (INS) del Perù a causa di vomito, diarrea, acidosi metabolica, ipokaliemia e iperlattatemia, ipoattività e febbre. A causa di questi sintomi, era stato precedentemente ricoverato all’età di 1 anno e 10 mesi e 2 anni e 2 mesi [1].
Il bambino è nato con un peso alla nascita di 3620 grammi, un’altezza di 49 cm e una circonferenza cranica di 34 cm (percentile normale) e un punteggio Apgar di 8-9. In termini di sviluppo psicomotorio, ha raggiunto il controllo della testa a un mese, si è seduto senza aiuto a sette mesi e ha camminato con sostegno a un anno e sei mesi. Ha pronunciato le prime parole a un anno e cinque mesi, ha detto due parole a due anni e nove mesi e ha mostrato un sorriso sociale a un anno. A 1 anno e 8 mesi di età, è stato valutato per il sottopeso e la crescita ridotta, la diarrea cronica, la febbre e l’aumento del volume addominale (Fig. 1).
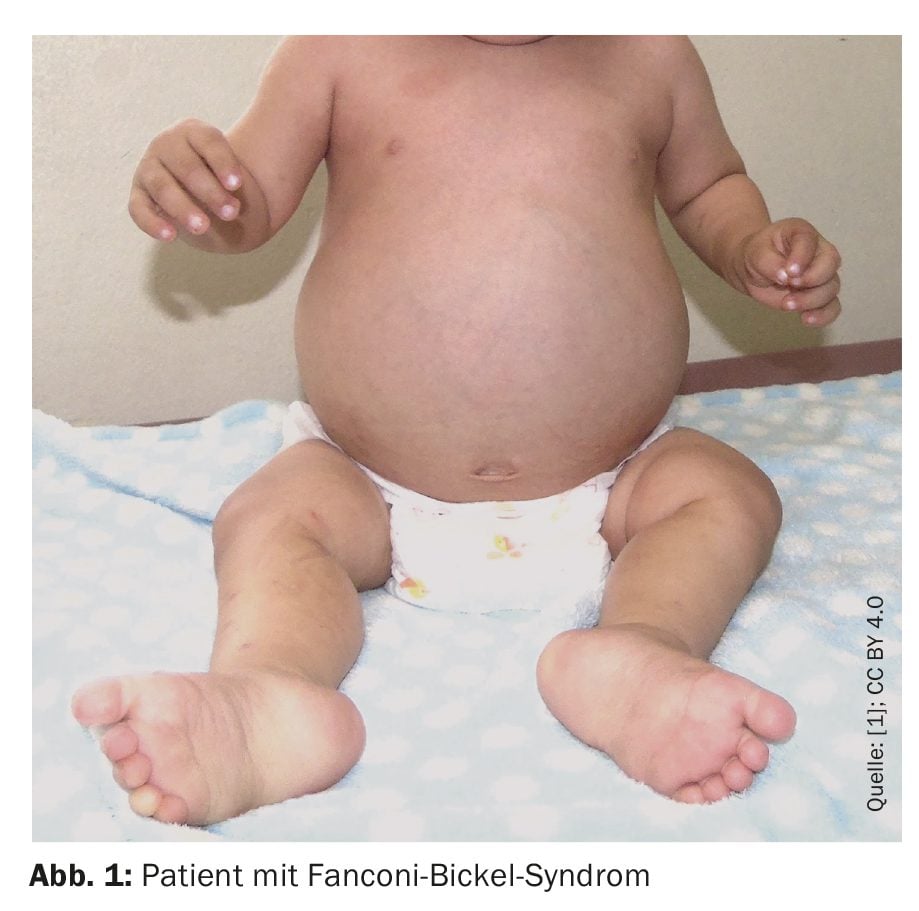
Al momento del ricovero, i medici hanno diagnosticato un rigonfiamento della fronte, epatomegalia, ipotonia e una deformità pseudo-madelungica. Il peso e l’altezza del bambino erano inferiori al primo percentile da quando aveva sei mesi, mentre la circonferenza della testa era nella norma. Le radiografie mostravano uno sfilacciamento e un allargamento delle metafisi del femore (distale) e della tibia (prossimale), coerenti con il rachitismo.
I risultati clinici e di laboratorio suggeriscono che la FBS
Il giovane paziente presentava ipoglicemia, ipo- e ipercalcemia, ipofosfatemia, ipercolesterolemia, ipertrigliceridemia, iperfosfatemia, ipokaliemia con diarrea e vomito, iperlattatemia e ipouricemia. Le analisi delle urine hanno mostrato valori normali di pH e HCO3, ma iperproteinuria, ipocreatinuria, microalbuminuria, iperglicosuria e ipercalciuria. Il bambino presentava anche trombocitosi. Il gas venoso mostrava un pH di 7,261-7,5 mmHg, HCO3 di 7,5-28,8 mmHg e un eccesso di base da -15,8 a +5,3. Queste analisi hanno portato alla diagnosi di acidosi tubulare renale. L’ecografia addominale mostrava epatomegalia e non c’erano segni di fibrosi o nefromegalia.
Una biopsia epatica ha rivelato un’architettura epatica parzialmente distorta per la presenza di un allargamento fibroso dello spazio portale, un’infiltrazione infiammatoria di linfociti, epatociti grandi e gonfiati con un modello a mosaico e una lieve fibrosi pericellulare. La colorazione periodica acido-Schiff con diastasi evidenzia depositi eosinofili negli epatociti che si correlano alla deposizione di glicogeno. Sulla base di questi risultati clinici e di laboratorio, i medici hanno sospettato la sindrome di Fanconi-Bickel (FBS).
Il sequenziamento dell’esoma ha identificato la variante patogena omozigote
I risultati dei test genetici sono stati ottenuti a 1 anno e 8 mesi di età utilizzando il DNA genomico. Sono state identificate 131.477 varianti annotate in 18.179 geni, escludendo le varianti probabilmente benigne o lievi. Per identificare le varianti associate al fenotipo del paziente, sono stati utilizzati i termini “mancata crescita” (HPO: 0001508) e “epatomegalia” (HPO: 0002240); gli autori hanno notato che è stata considerata una soglia di frequenza allelica della popolazione dell’1%. A causa dell’ipofosfatemia, del rachitismo e dell’accumulo di glicogeno nel fegato, i ricercatori hanno anche cercato manualmente le varianti in SLC2A2. A causa della consanguineità dei genitori, è stata data priorità all’analisi degli omozigoti. Una frequenza allelica delle varianti (VAF) superiore a 0,9 è stata utilizzata per selezionare i potenziali geni candidati. Il sequenziamento dell’esoma ha identificato una variante omozigote nonsense nel gene SLC2A2, che è stata descritta come patogena.
La diagnosi clinica del paziente si basava sulla presenza di epatomegalia, ipoglicemia, glucosuria, ipofosfatemia, iperfosfatemia, ipouricemia, rachitismo, deformità pseudo-madelungica e acidosi tubulare renale. Tuttavia, non è stato possibile determinare la presenza di aminoaciduria, poiché il test non era disponibile in loco. Questa insufficiente escrezione di aminoacidi avrebbe facilitato la diagnosi clinico-biochimica, spiegano gli autori. La conferma molecolare mediante il sequenziamento Sanger del gene SLCA2 ha rappresentato un altro ostacolo, poiché questo test non è disponibile in Perù. Dato l’accesso limitato ai test genetici nel Paese, non è stato possibile confermare la presenza della variante nei parenti di primo grado della paziente, sottolineano gli autori. In questo caso, il sequenziamento dell’esoma ha permesso ai ricercatori di identificare con precisione la variante omozigote.
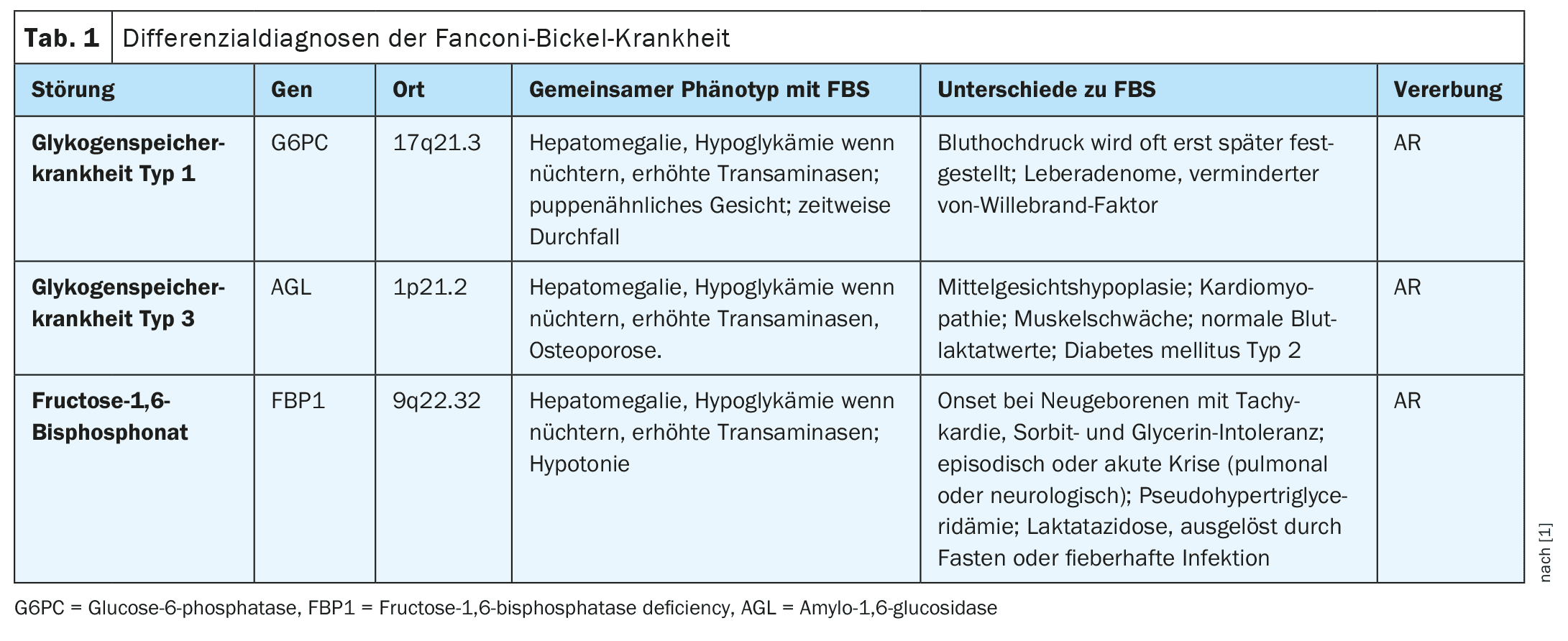
Secondo il dottor Abarca-Barriga e colleghi, l’aumento osservato del lattato nel sangue potrebbe essere dovuto a un maggiore carico di glucosio dal metabolismo anaerobico. Inoltre, un basso livello di acido urico dovuto alla disfunzione del tubulo prossimale fa parte del fenotipo dei pazienti con FBS, che porta all’iperuricosuria. Alcune caratteristiche cliniche, come il ritardo nello sviluppo del linguaggio, sono probabilmente associate alla presenza di ipoglicemia cronica.
Attualmente non esiste una terapia causale per la sindrome di Fanconi-Bickel. Il giovane paziente è stato trattato con bicarbonato, amlodipina, soluzione di sodio citrato e acido citrico, enalapril, alendronato e zolendronato e ha ricevuto un trattamento dietetico con amido di mais non cotto, che ha portato a un miglioramento del peso e dell’altezza. Inoltre, gli autori sottolineano che il paziente ha mostrato un miglioramento di 1 deviazione standard (SD) nel peso e nell’altezza da quando ha iniziato il trattamento dietetico con amido di mais non cotto. Pertanto, l’uso dell’amido di mais è essenziale non solo per prevenire l’ipoglicemia notturna, ma anche per migliorare l’altezza e il peso del paziente.
Letteratura:
- Abarca-Barriga HH, et al.: Importance about use of high-throughput sequencing in pediatric: case report of a patient with Fanconi-Bickel syndrome. BMC Pediatr 2024; 24: 161; doi: 10.1186/s12887-024-04641-1.
HAUSARZT PRAXIS 2024; 19(11): 48–49









