Per la prognosi di questa malattia ereditaria autosomica recessiva, la diagnosi nel periodo neonatale e il successivo inizio del trattamento sono cruciali – in particolare per quanto riguarda l’insorgenza di complicazioni come il carcinoma epatocellulare e il deterioramento neurocognitivo. Il nitisinone è attualmente disponibile come terapia farmacologica e si consigliano anche alcune misure dietetiche.
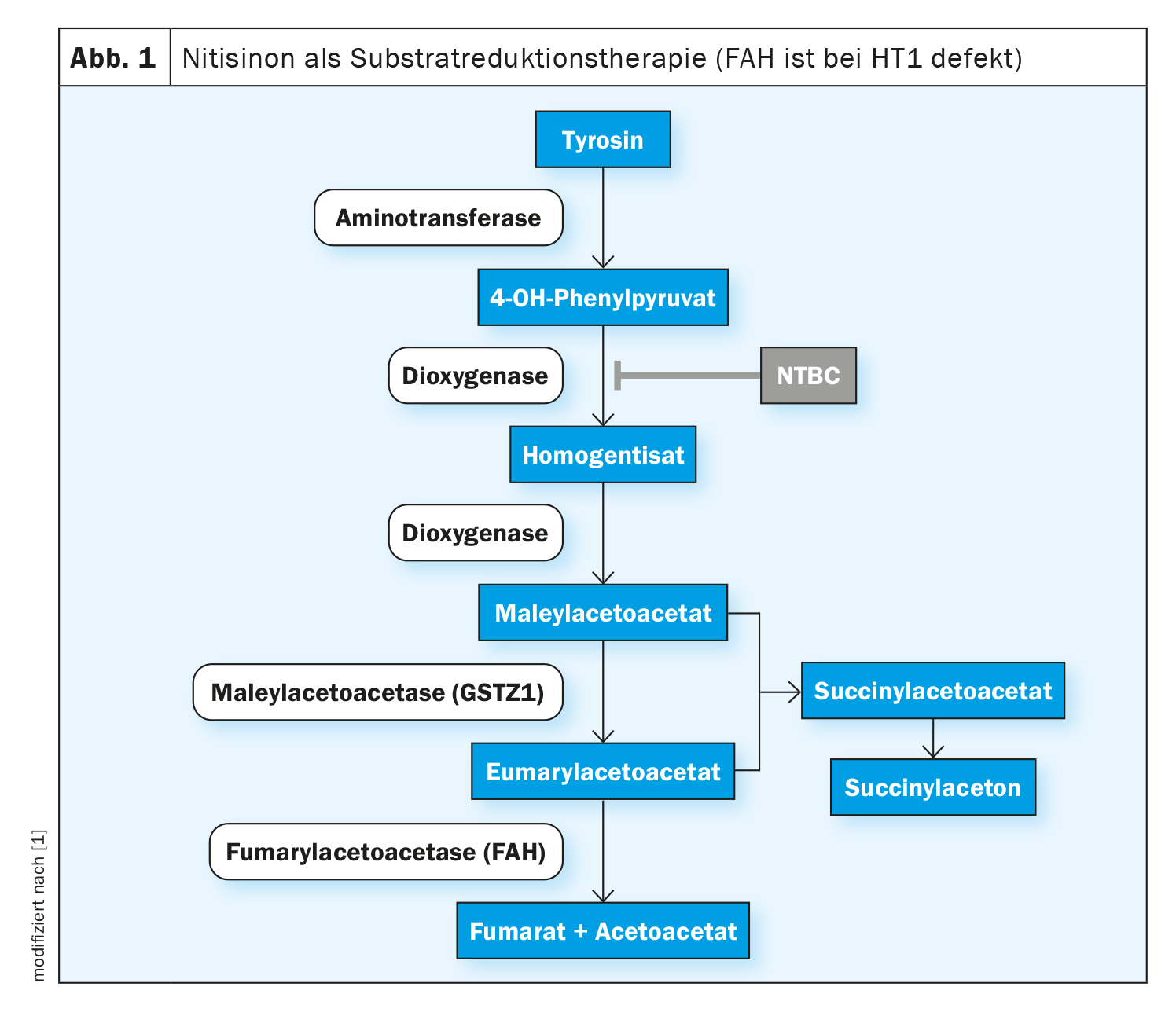
Nella tirosinemia epatorenale di tipo 1 (HT1), la formazione dell’enzima fumarilacetoacetasi (FAH), l’ultimo enzima nella degradazione della tirosina, è compromessa fin dalla nascita. Ciò è dovuto a una variante patogena biallelica nel gene FAH. A causa della carenza enzimatica, i metaboliti tossici fumarilacetoacetato, maleilacetoacetato e succinilacetone si accumulano nell’organismo e danneggiano fegato, reni e sistema nervoso periferico. Di conseguenza, l’insufficienza epatica non trattata si verifica spesso nel primo anno di vita o, con un decorso cronico più lento, si sviluppano la cirrosi epatica o il carcinoma epatocellulare (HCC) [1]. Il nitisinone è un principio attivo che interviene nella cascata di degradazione precoce della tirosina inibendo in modo competitivo l’enzima 4-idrossifenilpiruvato diossigenasi. Questo impedisce la formazione di prodotti intermedi tossici. Il trattamento di tutti i genotipi della malattia deve essere iniziato il più precocemente possibile, al fine di prolungare la sopravvivenza complessiva e prevenire le manifestazioni organiche pericolose per la vita. In Svizzera, il nitisinone (Nityr®) è stato autorizzato sotto forma di compresse nel 2022 [2]. Iniziare il trattamento nelle prime settimane di vita non solo previene l’HCC nella maggior parte dei casi, ma anche la disfunzione epatica e renale [3–5].
Prima dell’era del nitisinone, la storia naturale dell’HT1 era solitamente fatale, il 90% dei pazienti HT1 moriva nei primi due anni di vita e l’unica opzione era il trapianto di fegato [1]. La disponibilità del nitisinone (NTBC) ha quindi cambiato radicalmente il decorso clinico e l’esito delle persone affette da HT1, secondo gli autori della linea guida S2k sulla diagnosi e il trattamento della tirosinemia epatorenale (tirosinemia di tipo 1), aggiornata nel 2022 [1].
Si raccomanda lo screening neonatale
Il fattore decisivo per la prognosi a lungo termine, in particolare per la prevenzione del carcinoma epatocellulare, è l’inizio del trattamento nel periodo neonatale. Poiché i primi sintomi non compaiono nella maggior parte dei pazienti fino a diversi mesi di età, la diagnosi precoce non è possibile su basi puramente cliniche [3]. Pertanto, in caso di sospetto clinico, il succinilazetone (SA) deve essere determinato quantitativamente da sangue secco, siero/plasma e/o urina (“screening selettivo”). La determinazione della concentrazione di tirosina da sola ha una sensibilità e una specificità insufficienti e quindi non è raccomandata [1]. Un valore di misurazione SA superiore a un cut-off definito è considerato un risultato di screening positivo. Per confermare la diagnosi, si può effettuare un’analisi genetica molecolare del gene FAH (riquadro). Se il gene FAHè normale, la linea guida raccomanda di analizzare il gene GSTZ1 [1].
| Analisi genetiche molecolari L’identificazione di varianti patogene bialleliche nel genedella fumarilacetoacetasi (FAH)conferma la diagnosi di tirosinemia di tipo 1 [1]. Per questa analisi si può utilizzare il DNA delle cellule del sangue o di altri tessuti del corpo (tampone della guancia). È possibile anche l’analisi dell’RNA da cellule del sangue, campioni di biopsia epatica o fibroblasti in coltura. L’analisi del singolo gene dei 14 esoni codificanti del gene FAHnei pazienti con un chiaro quadro clinico/biochimico è un metodo consolidato che rileva le varianti patogene con una sensibilità stimata di >95% [1]. Al giorno d’oggi, tuttavia, le tecniche di NGS(Next Generation Sequencing) sono per lo più utilizzate come parte della diagnostica a pannelli o come parte del WES (Whole Exome Sequencing) per l’analisi del gene FAH– sia per una diagnostica più rapida che per una più conveniente [16]. |
Inizi la terapia con nitisinone nelle prime settimane di vita.
Gli effetti del nitisinone si basano sull’inibizione dell’enzima 4-idrossifenilpiruvato diossigenasi, che è coinvolto nella normale degradazione della tirosina (Fig. 1). Ciò impedisce la formazione di metaboliti tossici. Gli effetti collaterali sono rari, i più frequentemente segnalati sono disturbi dell’emocromo, disturbi oculari e un aumento della concentrazione di tirosina. La sospensione o l’interruzione improvvisa della terapia con nitisinone può portare a “crisi di porfiria” e deve essere evitata [1, 6-8]. Il valore target terapeutico per il nitisinone non è ancora stato ben definito; nella letteratura specializzata, viene indicato 20-60 μM come valore target terapeutico per le concentrazioni di nitisinone nel plasma [3,9–11]. Secondo gli autori delle linee guida, sono possibili anche concentrazioni significativamente inferiori senza compromettere il controllo metabolico, misurato dalla soppressione della produzione di SA [1]. L’intervallo terapeutico target per la concentrazione di tirosina nel sangue è indicato come 200-800 μM, senza che vi siano studi controllati, randomizzati e comparativi su questo [3,9–12].
Se possibile, la terapia con nitisinone deve essere combinata con una dieta a ridotto contenuto proteico, integrata con una miscela di aminoacidi priva di tirosina e fenilalanina [13–15].
Insidie diagnostiche – le analisi genetiche possono essere d’aiuto
La diagnosi differenziale più importante di una concentrazione elevata di SA è il deficit di maleil acetoacetato isomerasi, un’anomalia metabolica presumibilmente benigna che non è associata a disfunzioni epatiche [1]. La linea guida sottolinea anche che le concentrazioni di SA leggermente elevate – a seconda del valore di cut-off utilizzato – possono anche essere temporanee o dovute a forme lievi della malattia che non richiedono un trattamento [1]. Un risultato di screening positivo deve essere confermato da uno o più metodi di analisi alternativi, oltre al controllo nello stesso materiale del campione (sangue secco). Questi includono l’analisi quantitativa della gascromatografia/spettrometria di massa (GC/MS) degli acidi organici nelle urine e la determinazione della SA nel sangue. Se c’è un forte sospetto, è consigliabile controllare anche i parametri di funzionalità epatica. I risultati anomali nella diagnostica di conferma possono essere ulteriormente chiariti dal test genetico molecolare del gene FAH. Se questo non è evidente, si può analizzare il gene GSTZ1 (glutatione S-transferasi zeta 1-1), la cui carenza è la causa del deficit di maleil acetoacetato isomerasi [1].
Letteratura:
- “Linea guida S2k: Diagnosi e trattamento della tirosinemia epatorenale (tirosinemia di tipo 1)”, numero di registro AWMF: 027-003, al 09.06.2022.
- Swissmedic: Informazioni sui medicinali, www.swissmedicinfo.ch,(ultimo accesso 06.02.2024)
- Mayorandan S, et al: Studio trasversale di 168 pazienti con tirosinemia epatorenale e implicazioni per la pratica clinica. Orphanet J Rare Dis 2014; 9: 107.
- McKiernan PJ, Preece MA, Chakrapani A: Esito dei bambini con tirosinemia ereditaria dopo lo screening neonatale. Arch Dis Child 2015; 100(8): 738-741.
- Bartlett DC, et al: Il trattamento precoce con nitisinone riduce la necessità di trapianto di fegato nei bambini con tirosinemia di tipo 1 e migliora la funzione renale post-trapianto. J Inherit Metab Dis 2014; 37(5): 745-752.
- Önenli Mungan N, et al: Tirosinemia di tipo 1 e crisi neurologica irreversibile dopo un mese di sospensione del nitisone. Metab Brain Dis 2016; 31(5): 1181-1183.
- Schlump JU, et al: Grave crisi neurologica in un paziente con tirosinemia ereditaria di tipo I dopo l’interruzione del trattamento con NTBC. J Inherit Metab Dis 2008; 31 Suppl 2: S223-S225.
- Uçar HK, et al: Un caso di associazione molto rara di tirosinemia di tipo I e pancreatite che imita la crisi neurologica della tirosinemia di tipo I. Balkan Med J 2016; 33(3): 370-372.
- Chinsky JM, et al: Diagnosi e trattamento della tirosinemia di tipo I: revisione e raccomandazioni del gruppo di consenso statunitense e canadese. Genet Med 2017; 19(12): doi:10.1038/gim.2017.101
- de Laet C, et al: Raccomandazioni per la gestione della tirosinemia di tipo 1. Orphanet J Rare Dis 2013; 8: 8. Pubblicato nel 2013 Jan 11. doi:10.1186/1750-1172-8-8
- Alvarez F, Mitchell GA: Tirosinemia e trapianto di fegato: esperienza al CHU Sainte-Justine. Adv Exp Med Biol 2017; 959: 67-73.
- De Laet C, et al: Esito neuropsicologico dei pazienti con tirosinemia di tipo 1 trattati con NTBC. Dev Med Child Neurol 2011; 53(10): 962-964.
- Morrow G, Angileri F, Tanguay RM: Aspetti molecolari delle mutazioni FAH coinvolte nella malattia HT1. Adv Exp Med Biol 2017; 959: 25-48.
- Morrow G, Tanguay RM: Aspetti biochimici e clinici della tirosinemia ereditaria di tipo 1 Adv Exp Med Biol 2017; 959: 9-21.
- van Spronsen FJ, et al: Considerazioni dietetiche nella tirosinemia di tipo I. Adv Exp Med Biol 2017; 959: 197-204.
- Blackburn PR, et al: Tirosinemia silenziosa di tipo I senza tirosina o succinilacetone elevati associata a cirrosi epatica e carcinoma epatocellulare. Hum Mutat 2016; 37(10): 1097-1105.
MEDICINA GENERALE 2024; 19(2): 36-37