I linfomi cutanei sono un gruppo eterogeneo di malattie neoplastiche che derivano dalla proliferazione clonale dei linfociti nella pelle. In generale, i linfomi cutanei sono forme rare di cancro della pelle, con una prevalenza di 1:100.000 abitanti. La micosi fungoide (MF) è il linfoma cutaneo a cellule T (CTCL) più comune. Gli stadi iniziali della MF sono trattati principalmente per via topica con corticosteroidi, fototerapia o radioterapia locale. Dal 2008 esiste una classificazione uniforme dell’OMS dei linfomi cutanei primari (basata su EORTC/WHO). Viene fatta una distinzione tra linfomi a cellule T e B, che è anche rilevante dal punto di vista clinico e terapeutico. I linfomi cutanei hanno una prognosi e una terapia completamente diverse rispetto ai linfomi linfatici istologicamente comparabili.
I linfomi cutanei sono malattie neoplastiche della pelle che si sviluppano quando i linfociti degenerati iniziano a crescere in modo incontrollato. I linfomi dei linfonodi sono particolarmente noti. Possono svilupparsi in tutti gli organi del sistema linfatico, compresi milza, timo e tonsille. Se i linfociti della pelle degenerano, si formano dei linfomi cutanei. I linfomi cutanei sono linfomi non-Hodgkin. A seconda del tipo di linfocita che diventa neoplastico, si distingue tra linfomi cutanei a cellule T e B (tab. 1).

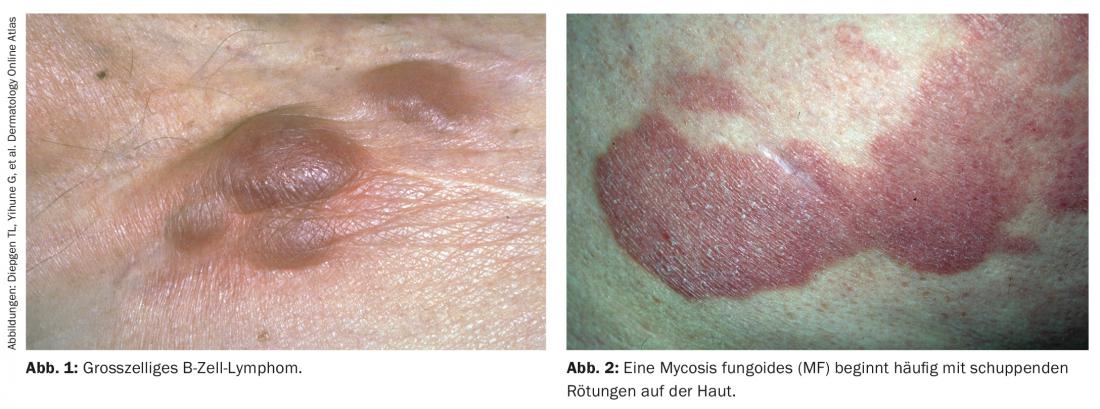
Questa classificazione ha anche molto senso, in quanto i linfomi a cellule T e B differiscono in modo significativo l’uno dall’altro, sia nel comportamento clinico che nella prognosi. I linfomi a cellule T sono generalmente più aggressivi dei linfomi a cellule B e quindi hanno una prognosi peggiore. I due tipi di linfoma cutaneo differiscono anche nell’aspetto: i linfomi a cellule T di solito appaiono generalizzati su tutto il corpo, mentre i linfomi a cellule B spesso appaiono come tumori solitari (Fig. 1) [1].
Linfomi cutanei a cellule T e sottotipi
Il linfoma cutaneo a cellule T più comune è la micosi fungoide (MF), oggi difficilmente conosciuta come “lichene da usura”. Il nome fuorviante di micosi fungoide fu introdotto nel 1832 da Jean-Louis-Marc Alibert, partendo dal presupposto che si trattasse di una malattia fungina. Tuttavia, l’eziologia della micosi fungoide e di tutti gli altri linfomi cutanei non è ancora chiara. Le scottature solari, come nel caso del melanoma, non sono una possibile causa di linfomi cutanei. Solo le alte dosi di radiazioni radioattive sono state confermate come fattore di rischio fino ad oggi. Il danno al materiale genetico dei linfociti causato dalle radiazioni porta alla degenerazione della divisione cellulare e alla proliferazione clonale delle cellule. Tuttavia, i linfociti possono anche degenerare senza danni esterni, perché sono cellule che si dividono rapidamente. Gli errori di lettura spontanei nel materiale genetico possono portare a linfomi [1].
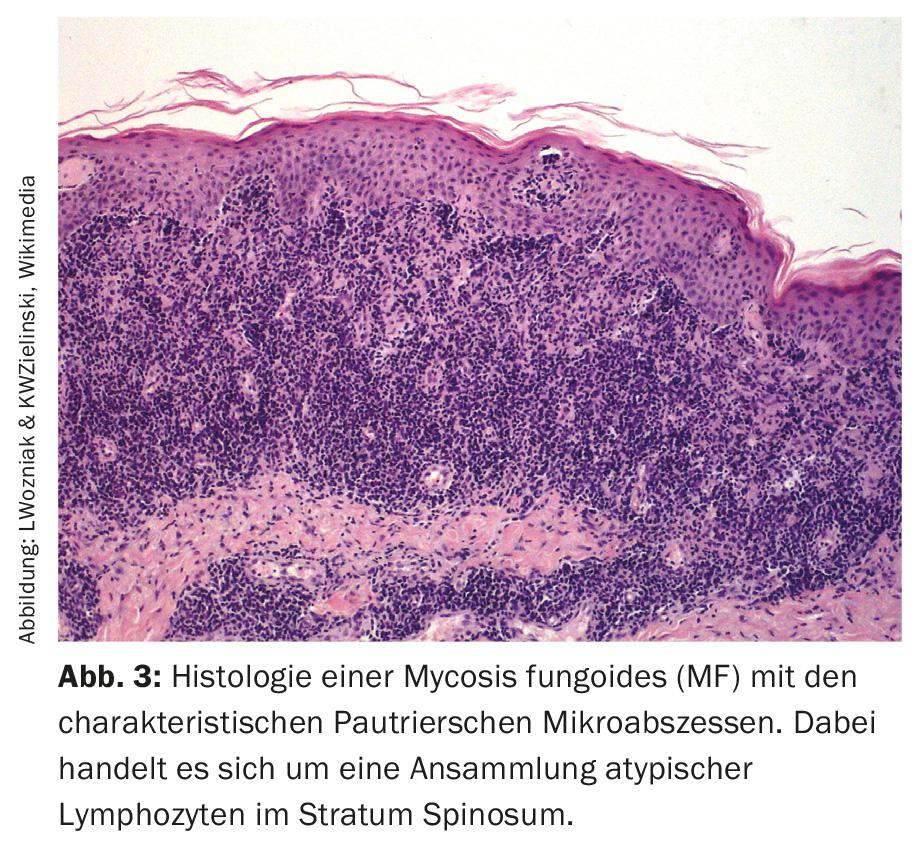
Micosi fungoide (MF): spesso mostra un decorso clinico tipico a fasi. La fase iniziale mostra un lento e progressivo stadio di chiazze simili a un eczema (Fig. 2). Clinicamente, i cambiamenti appaiono come alterazioni cutanee eczematose. Questo stadio può persistere per anni e può poi passare allo stadio di placca (Fig. 4). In questa fase, le lesioni sono palpabili e ispessite. Inoltre, lo stadio di placca può trasformarsi in uno stadio tumorale, spesso molto impressionante dal punto di vista clinico. Tuttavia, queste fasi non devono necessariamente susseguirsi una dopo l’altra nel modo classico. Ad esempio, si può osservare una transizione dallo stadio di chiazza direttamente allo stadio di tumore o alla forma eritrodematosa, la cosiddetta sindrome di Sézary (SS) (Fig. 5). La cosiddetta variante leucemica della micosi fungoide mostra una leucocitosi tra 10.000-50.000 (raramente fino a 100.000 leucociti/ml). Si tratta di un quadro clinico serio.
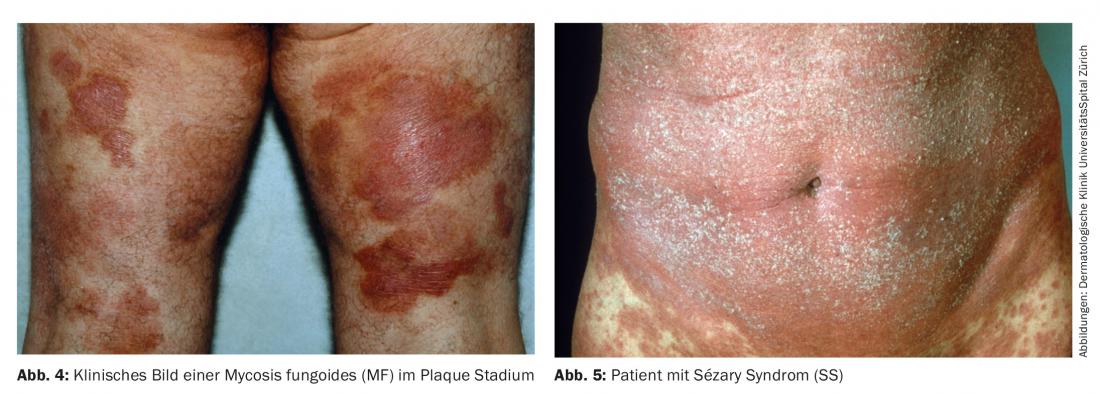
Sindrome di Sézary: è intesa come la variante leucemica della micosi fungoide e ha anche una prognosi significativamente peggiore rispetto alla micosi fungoide (Tab. 5). Le cosiddette cellule di Sézary (linfociti atipici) si trovano nel sangue periferico. Inoltre, in questi pazienti l’ingrossamento dei linfonodi è palpabile e l’eritroderma è clinicamente evidente. Il paziente spesso sperimenta un grave prurito e le condizioni generali sono di solito significativamente ridotte.

Linfoma cutaneo a cellule T CD30+ a grandi cellule: istologicamente, il linfoma cutaneo a cellule T CD30+ a grandi cellule corrisponde a quello del linfoma nodale dello stesso tipo. Tuttavia, la prognosi è diametralmente diversa e quindi deve essere trattata terapeuticamente in modo diverso. La prognosi dei linfomi nodali di questo tipo è molto sfavorevole e richiede una chemioterapia costosa. Nel linfoma a grandi cellule T CD30+ (pleomorfo o anaplastico) della pelle, tuttavia, la prognosi è eccellente e la chemioterapia è controindicata.
Linfomi cutanei a cellule B
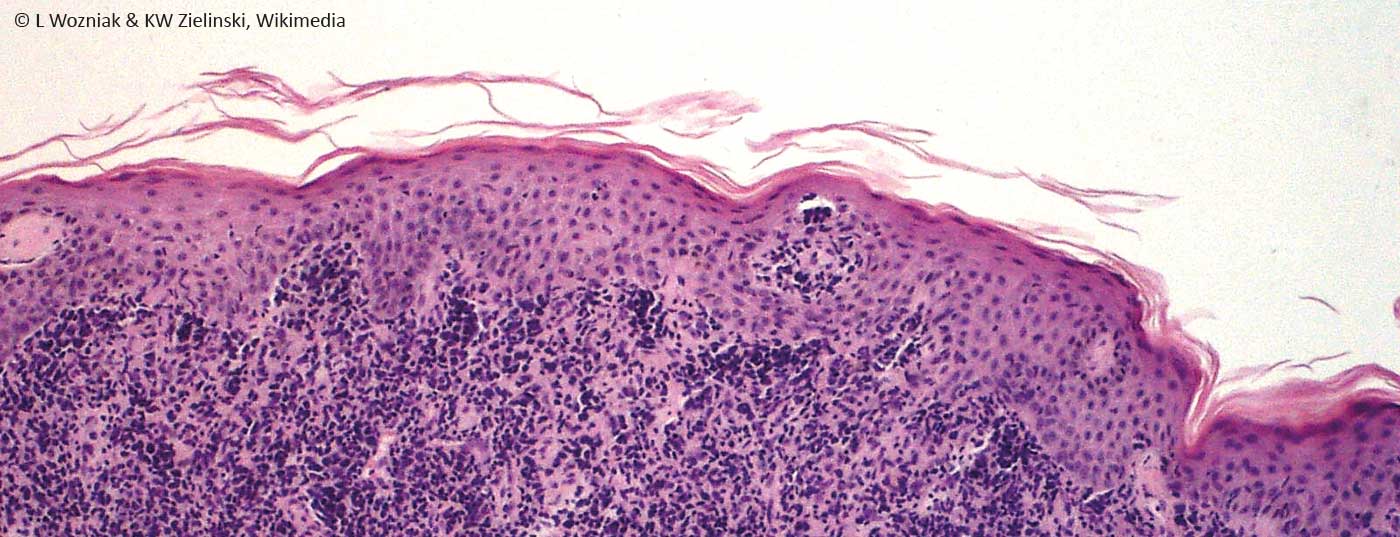
Il raro linfoma a cellule B (circa il 20% di tutti i linfomi cutanei) è solitamente un tumore solitario, a crescita rapida. Le manifestazioni extracutanee sono molto rare e quindi la prognosi è spesso eccellente. I noduli sono per lo più noduli indolenti e lisci, che di solito si presentano in modo solitario, a volte anche disseminati. Sono di colore rosso, in parte brunastro. Spesso c’è un infiltrato dermico, a volte sottocutaneo con epidermide intatta. Il quadro istologico dei linfomi cutanei a cellule B è piuttosto monomorfico, a differenza dei linfomi a cellule T, che mostrano una grande diversità istologica (Fig. 3) [1].
Stadi del linfoma a cellule T
Per la selezione di una terapia adeguata, è necessario riconoscere non solo il tipo, ma anche lo stadio in cui si trova il linfoma cutaneo (Tab. 2). Lo stadio fornisce informazioni sul grado di diffusione delle cellule tumorali in altre parti del corpo. Nello stadio I, solo alcune parti della pelle sono colpite dal linfoma. La pelle presenta chiazze squamose e arrossate, ma non noduli. Nello stadio II, sono visibili anche ispessimenti nodulari sulla pelle, oppure i linfonodi sono ingrossati. Nello stadio III, il linfoma cutaneo copre quasi tutta la pelle. Il criterio per l’ultimo stadio IV è l’infestazione di altri organi con cellule tumorali. Possono essere colpiti i linfonodi o altri organi.

In genere, i linfomi cutanei si manifestano nei pazienti di età superiore ai 20 anni. I linfomi cutanei ad alta malignità mostrano un picco di età tra i 5-15 anni e i 60-70 anni [5].
Diagnostica
I linfomi cutanei sono complessivamente rari e spesso assomigliano clinicamente ad altre malattie dermatologiche. Pertanto, lo stadio primario della MF è spesso confuso e simile alla dermatite da contatto, cosicché il medico viene spesso avvisato del linfoma cutaneo solo dal decorso clinicamente persistente. Queste neoplasie cutanee devono essere sottoposte a biopsia. Oggi è possibile il rilevamento monoclonale dei linfociti a partire dalla preparazione istologica. Questa clonalità può essere rilevata a livello del DNA, dell’mRNA e delle proteine, per cui si possono individuare i geni dei recettori delle cellule T o delle immunoglobuline [1].
Esame clinico
L’esame clinico per il sospetto di linfoma cutaneo comprende una valutazione dettagliata di tutte le lesioni cutanee, nonché uno stato linfonodale di tutte le stazioni linfonodali (palpazione delle stazioni linfonodali, fegato, milza). Le procedure di imaging includono l’ecografia addominale e dei linfonodi e le radiografie del torace. L’esame dipende dallo stadio della malattia del linfoma a cellule T cutaneo. Per esempio, la TAC dell’intero corpo per il linfoma a cellule T viene eseguita solo a partire dallo stadio IIB in caso di micosi fungoide, sindrome di Sézary (SS), leucemia/linfoma a cellule T dell’adulto, disordini linfoproliferativi primari CD30+, linfoma a cellule T sottocutaneo simile alla panniculite, linfoma extranodale a cellule NK/T (tipo nasale) e linfoma a cellule T cutaneo primario (non specificato).
Nei linfomi a cellule B, la TAC dell’intero corpo è indicata per il linfoma primario cutaneo diffuso a grandi cellule B (tipo gamba) (PCBLT) e (PCBLT di altri tipi), oltre che per il linfoma primario cutaneo intravascolare a grandi cellule B.
La diagnosi di linfomi cutanei deve sempre essere confermata istologicamente mediante una biopsia. Gli esami immunoistochimici e la possibilità di esami biologici molecolari, con cui si può esaminare la clonalità dei linfociti, sono spesso molto utili per la differenziazione da altre malattie infiammatorie della pelle.
Test di laboratorio
Se viene rilevato un linfoma cutaneo, si raccomanda di eseguire i seguenti esami di laboratorio:
- VES e CRP, emocromo, enzimi epatici, creatinina, LDH, elettroliti.
- Se necessario: immunoelettroforesi, sierologia HTLV (soprattutto per i pazienti provenienti dall’estero), sierologia Borrelia, esami ematologici speciali, ulteriori esami di laboratorio a seconda della terapia prevista.
- Nei linfomi eritrodermici a cellule T (sospetta sindrome di Sézary), è indicato uno striscio di sangue per le cellule di Sézary.
Per i linfomi a cellule B, possono essere necessarie ulteriori biopsie del midollo osseo: obbligatorie per il PCBCL, facoltative per il PCMZL e il PCFCL.
Stadi TNM
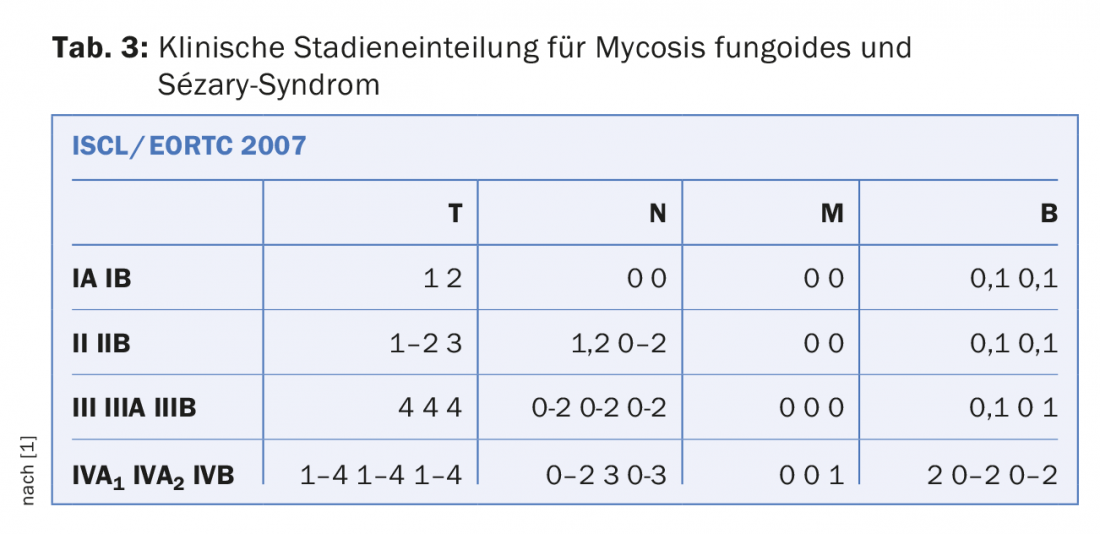
La stadiazione dei linfomi cutanei CTCL viene effettuata secondo la classificazione TNM (Tab. 3). La classificazione fornisce informazioni sulla prognosi e quindi indirettamente anche sulla terapia necessaria. Non si sottolineerà mai abbastanza che i linfomi cutanei e i linfomi nodali hanno una clinica completamente diversa e devono quindi essere valutati in modo del tutto indipendente (nonostante l’istologia e la classificazione talvolta quasi identiche). In passato, spesso i linfomi cutanei (CTCL) venivano trattati in modo eccessivo. Spesso sono stati utilizzati regimi terapeutici identici a quelli dei linfomi nodali, che non solo sono inefficaci, ma anche dannosi per il paziente: I linfomi cutanei hanno una prognosi e una terapia completamente diverse rispetto ai linfomi linfatici istologicamente comparabili! Pertanto, anche la classificazione TNM non è comparabile 1:1 con i linfomi nodali.
Previsioni
Gli stadi precoci della MF (IA-IIA) hanno solitamente una prognosi molto buona. Il tempo medio di sopravvivenza è di 10-20 anni. I linfomi cutanei possono essere suddivisi in tre classi prognostiche:
1. linfomi cutanei con buona prognosi:
- Micosi fungoide (MF)
- Reticolosi pagetoide
- Granulomatosi della pelle lassa
- Linfoma cutaneo primario a cellule T pleomorfe medio-piccole (provvisorio)
- Linfoma anaplastico a grandi cellule primario cutaneo
- Papulosi linfomatoide
- Linfoma a cellule T sottocutaneo simile alla pannicolite
- Linfoma primario cutaneo a cellule B della zona marginale
- Linfoma follicolare primario cutaneo
2. linfomi cutanei con prognosi moderata:
- Follicolotropico MF
- Sindrome di Sézary
- Leucemia/linfoma a cellule T dell’adulto (HTLV+)
- Linfoma primario cutaneo diffuso a grandi cellule B, tipo gamba/altri tipi
3. linfomi cutanei con prognosi sfavorevole
- Linfoma extranodale a cellule NK/T (tipo nasale)
- Linfoma cutaneo primario aggressivo epidermotropico a cellule T CD8+ (provvisorio)
- Linfoma cutaneo a cellule T (provvisorio)
- Linfoma cutaneo primario intravascolare a grandi cellule B
- Linfoma a cellule T sottocutaneo simile alla pannicolite con emofagocitosi
- Neoplasia ematoderma CD4+, CD56+
Terapia
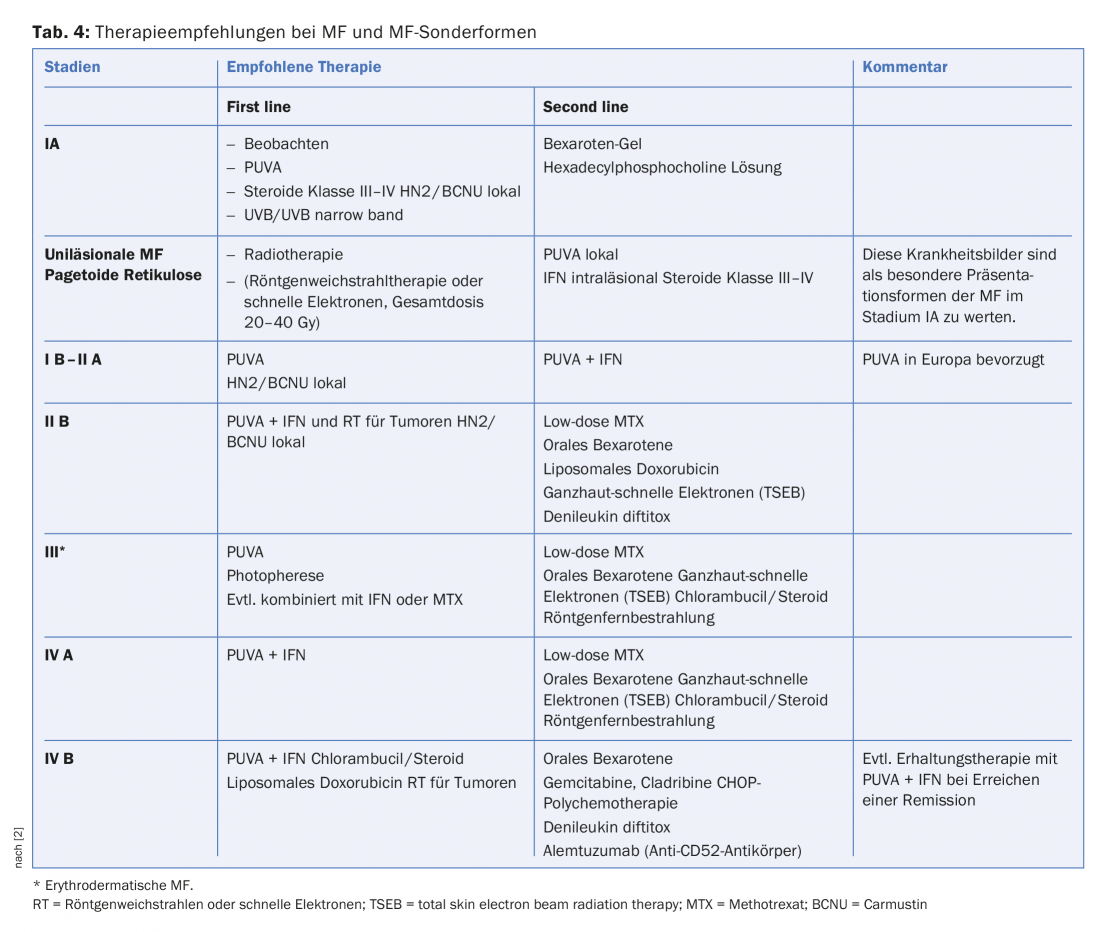
Terapia dei linfomi cutanei a cellule T: le fasi iniziali della micosi fungoide, come lo stadio eczematoso angolare e in parte anche lo stadio a chiazze, possono spesso essere trattate con successo per anni con steroidi locali (talvolta combinati con PUVA o luce UVB a banda stretta). Se non c’è risposta, si utilizza l’interferone come prima terapia di sistema. Le terapie per gli stadi più elevati dei linfomi cutanei sono riassunte nella tabella 4 .
Terapia dei linfomi cutanei a cellule B (CBCL)
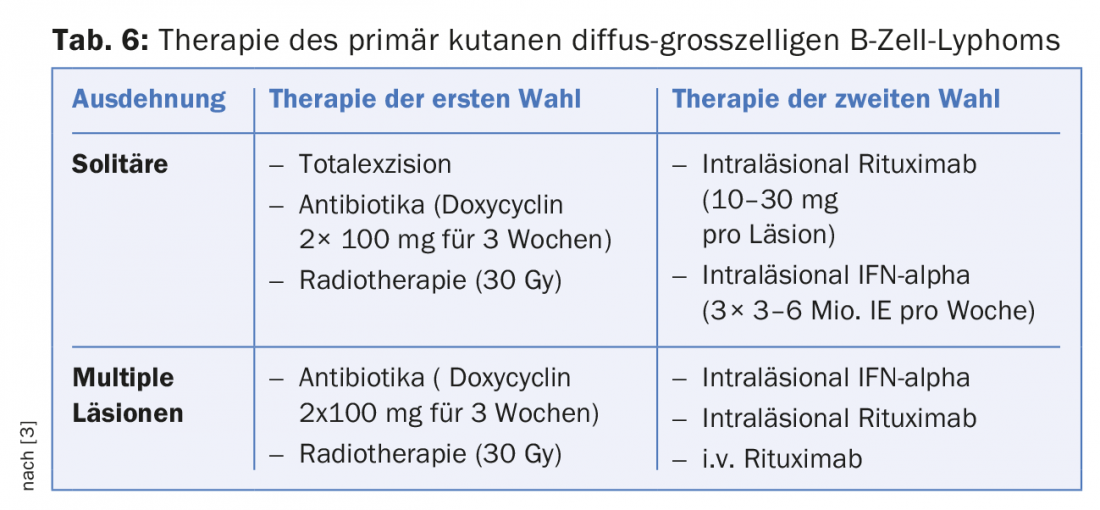
Linfomi primari cutanei a cellule B della zona marginale e linfomi del centro germinale: i linfomi cutanei a cellule B poco maligni corrispondono ai linfomi MALT nodali (tessuto linfoide associato alla mucosa) a causa della loro somiglianza morfologica. A causa delle somiglianze, i CBCL a bassa malignità sono chiamati da alcuni autori linfomi SALT (tessuto linfoide associato alla pelle). La prognosi di questo gruppo di malattie è generalmente estremamente favorevole. Poiché in alcuni casi è possibile rilevare particelle infettive (DNA di Borrelia), inizialmente si raccomanda il trattamento con un antibiotico ad ampio spettro. Poiché la rilevazione della Borrelia può essere falsamente negativa ed è anche costosa, ai pazienti viene raccomandata una terapia di tre settimane con doxiciclina al dosaggio di 2× 100 mg p.o. al giorno.
L’escissione chirurgica è consigliata per le lesioni piccole, mentre la radioterapia deve essere presa in considerazione per quelle più grandi. Si raccomanda una dose totale di 20 Gy-35 Gy, frazionata in dosi singole, da 1 a 2× 4 Gy, poi 2 Gy fino alla dose totale, da 2 a 3× alla settimana. In alternativa, si consigliano iniezioni intralesionali di interferone o l’uso di rituximab, un anticorpo umanizzato diretto contro l’antigene delle cellule B CD20. La terapia intralesionale è possibile anche con questa sostanza, per cui è necessario utilizzare solo il 20% della dose necessaria a livello sistemico.
A differenza dei linfomi cutanei a cellule B con struttura follicolare, i linfomi del centro germinale e i linfomi diffusi a grandi cellule B presentano una prognosi relativamente scarsa. La radioterapia è consigliata per prima, con dosi di almeno 30Gy. In caso di recidive, si può prendere in considerazione la polichemioterapia combinata con rituximab [3].
Aspetti psico-oncologici
I pochi studi sul benessere psicologico e sociale dei pazienti affetti da linfoma cutaneo mostrano una drastica riduzione della qualità di vita. Soprattutto il trattamento con steroidi e interferone sembra scatenare stati depressivi e ansia in alcuni pazienti. I pazienti con MF hanno descritto principalmente la loro stanchezza e i disturbi del sonno che si verificano come fattori limitanti. Anche le difficoltà nel rapporto con gli altri, nel lavoro e nelle relazioni personali sono molto stressanti a causa della ‘visibilità’ della diagnosi [1].
Prospettiva
Un nuovo approccio terapeutico per i linfomi cutanei potrebbe essere la terapia fotodinamica (PDT). Questa terapia è già utilizzata con successo nel trattamento delle cheratosi attiniche e della malattia di Bowen. Il gruppo del Professor Hunger a Berna ha intrapreso una sperimentazione pilota su pazienti con MF nelle prime fasi della malattia. I risultati hanno mostrato che è stata ottenuta una risposta nel 73% delle lesioni trattate. Il gruppo conclude che i pazienti con stadi precoci di MF possono essere trattati con successo con la PDT, con risultati cosmetici eccellenti [4].
Letteratura:
- www.derma.de/de/daten/leitlinien/leitlinien/kutane-lymphome
- www.awmf.org/uploads/tx_szleitlinien/032-027l_Kutane_Lymphome_2014-verlaengert.pdf
- medicalforum.ch/docs/smf/archiv/de/2009/2009-42/2009-42-379.pdf
- Derm. Hel. 2016;28(7): 28-32
- www.dermatologie.usz.ch/ueber-die-klinik/Documents/USZ_Lymphom_A5.pdf
PRATICA DERMATOLOGICA 2016; 26(6): 6-12