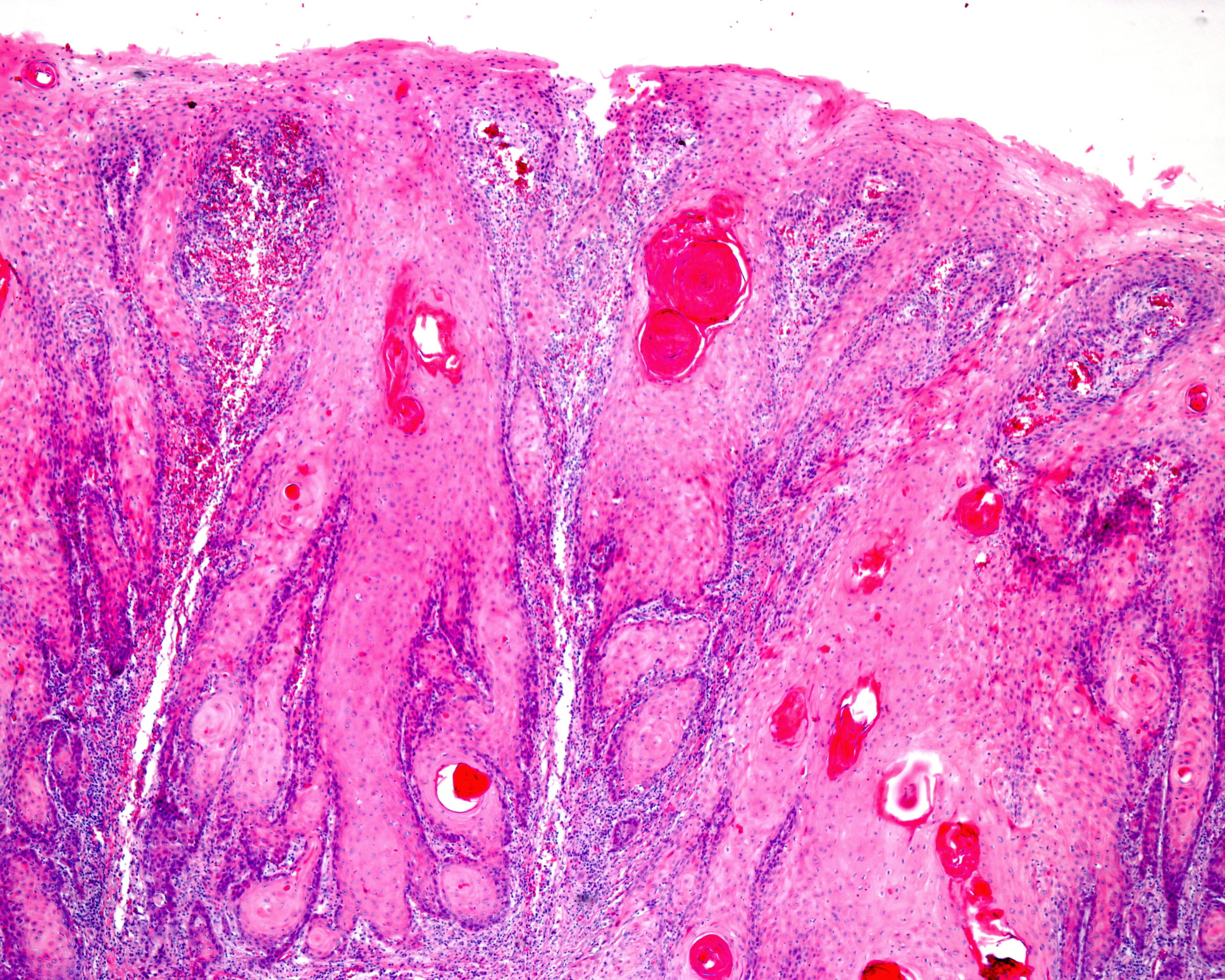
As amiloidoses são doenças de desdobramento proteico em que há normalmente deposição extracelular de fibrilas proteicas insolúveis com uma configuração antiparalela da folha β. Os patomecanismos subjacentes são múltiplos. As causas adquiridas são, por exemplo, concentrações elevadas das proteínas em questão no sangue ou tecido de uma doença ou inflamação clonal subjacente.
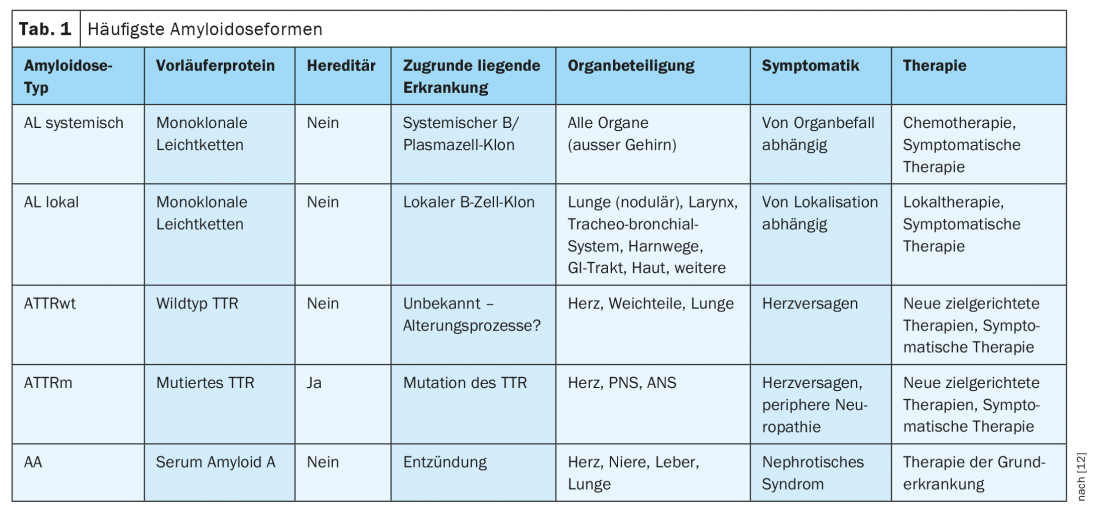
As amiloidoses são doenças de desdobramento proteico em que há normalmente deposição extracelular de fibrilas proteicas insolúveis com uma configuração antiparalela da folha β[1]. No homem, são actualmente conhecidas 36 proteínas amiloidogénicas que podem ser depositadas sob a forma destes agregados [2]. Os patomecanismos subjacentes são múltiplos. As causas adquiridas são, por exemplo, concentrações elevadas das proteínas em questão no sangue ou tecido de uma doença ou inflamação clonal subjacente. As amiloidoses hereditárias, por outro lado, baseiam-se principalmente em mutações pontuais com uma estrutura terciária da proteína em causa posteriormente alterada. Os agregados resultantes podem ser locais (local idêntico de produção e deposição) ou sistémicos (diferentes locais de produção e deposição) e mostram frequentemente o organismo tropismo característico do subtipo de amiloidose correspondente. A designação das formas de amiloidose é feita de acordo com a nomenclatura internacional por siglas, onde a letra maiúscula “A” é seguida por uma abreviatura para a respectiva proteína precursora [2]. As amiloidoses mais comuns e as suas características centrais estão resumidas no Quadro 1.
A amiloidose mais comum nos países industrializados é a amiloidose sistémica de AL. Com cerca de 10 casos por milhão de pessoas-ano [3], é uma das doenças raras. É geralmente causada por células plasma monoclonais na medula óssea que produzem uma cadeia ligeira amilóide. Os depósitos de amiloidose AL também são encontrados em muitos casos de amiloidose local. Em contraste com a amiloidose AL sistémica, porém, a amiloide AL é depositada localmente ou multilocularmente e está quase sempre limitada a um sistema de órgãos. Nestes casos, o plasma clonal ou as células B estão frequentemente presentes apenas localmente e uma gamopatia monoclonal não é ou é apenas muito ligeiramente detectável no sangue ou na urina [4]. Pouco se sabe sobre a génese da amiloidose AL local. Presumivelmente, a estimulação local das células B induzida por antigénios também desempenha um papel importante em muitos casos [5]. Além disso, o aumento da actividade das células B no contexto de uma doença reumatológica inflamatória subjacente pode ser causador [6,7].
Os sintomas e as opções de tratamento da respectiva amiloidose derivam da patologia subjacente e do envolvimento dos órgãos. O espectro varia desde a não necessidade de tratamento com uma esperança de vida e qualidade ilimitadas, até uma doença potencialmente ameaçadora mas facilmente tratável, até à doença mais grave com um prognóstico extremamente desfavorável [8,9]. Devido à raridade das amiloidoses e aos sintomas muitas vezes inespecíficos, o diagnóstico é infelizmente feito tardiamente, numa altura em que, no caso das amiloidoses sistémicas, já ocorreram frequentemente danos graves e dificilmente reversíveis dos órgãos terminais. As provas histológicas de material tipicamente birefringente no Congo, a coloração vermelha é obrigatória para confirmar o diagnóstico (excepto na amiloidose ATTR do coração). O tecido pode ser obtido a partir do órgão afectado ou, no caso de amiloidoses sistémicas, a partir de qualquer outro local potencial de deposição. Na amiloidose sistémica de cadeia ligeira (AL), a aspiração de tecido adiposo provou ser um método muito sensível para confirmar o diagnóstico [10]. Para o diagnóstico diferencial e classificação exacta da amiloidose, bem como para o desenvolvimento de uma estratégia de tratamento individual, recomenda-se que o paciente seja apresentado num centro de amiloidose. Em particular, a subtilografia histológica, a constelação clínica dos achados, bem como a extensão dos danos dos órgãos e, consequentemente, a capacidade terapêutica são tidas em conta. A norma para diferenciar as várias formas de amiloidose na Alemanha é a imuno-histoquímica, que deve ser realizada por patologistas ou institutos especializados [11]. Uma nova biópsia não é normalmente necessária.
Amiloidose pulmonar
A amiloidose pulmonar é um diagnóstico muito raro. No entanto, a prevalência exacta é desconhecida, uma vez que é frequentemente assintomática e, portanto, muitas vezes acidental ou mesmo post-mortem. De Abril de 2000 a Outubro de 2019, vimos um total de 157 doentes com amiloidose pulmonar local no nosso centro [6]. Durante o mesmo período, vimos 2240 pacientes com a amiloidose sistémica mais comum na Alemanha e na Suíça, a amiloidose AL. Estudos de autópsia sugerem que o depósito amiloide pulmonar e as alterações consecutivas do tecido pulmonar são detectáveis em até 90% da amiloidose AL sistémica [12]. Contudo, os sintomas clínicos dos pacientes afectados só muito raramente são atribuídos ao envolvimento pulmonar, mesmo quando estão presentes anomalias morfológicas por TC [13]. Isto deve-se ao facto de que na maioria dos pacientes com amiloidose AL sistémica, o envolvimento cardíaco é clinicamente importante e pode ser facilmente confirmado pela ecocardiografia [9]. Os sintomas pulmonares isolados, por outro lado, são mais comuns na amiloidose AL local [6]. Estas também representam a maioria das amiloidoses pulmonares diagnosticadas ante mortem. A esperança de vida dos pacientes com amiloidose AL local não é geralmente limitada em comparação com a população em geral. Além disso, o risco de progressão para amiloidose AL sistémica é geralmente baixo. Contudo, o curso clínico é frequentemente caracterizado por recorrências ou progressões locais.
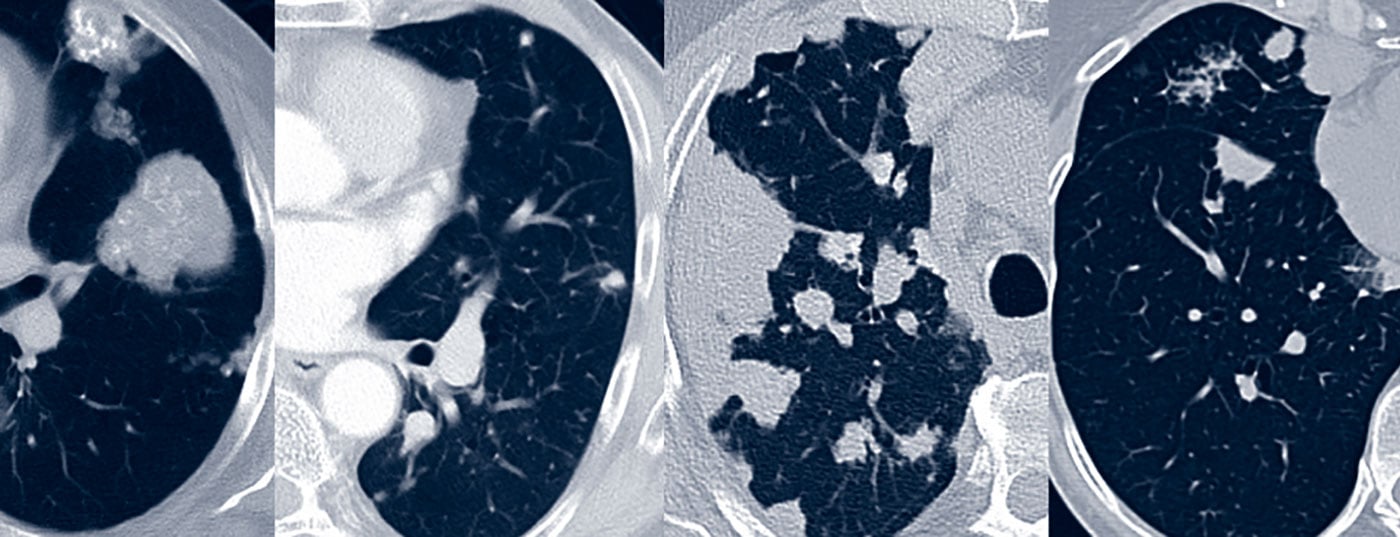
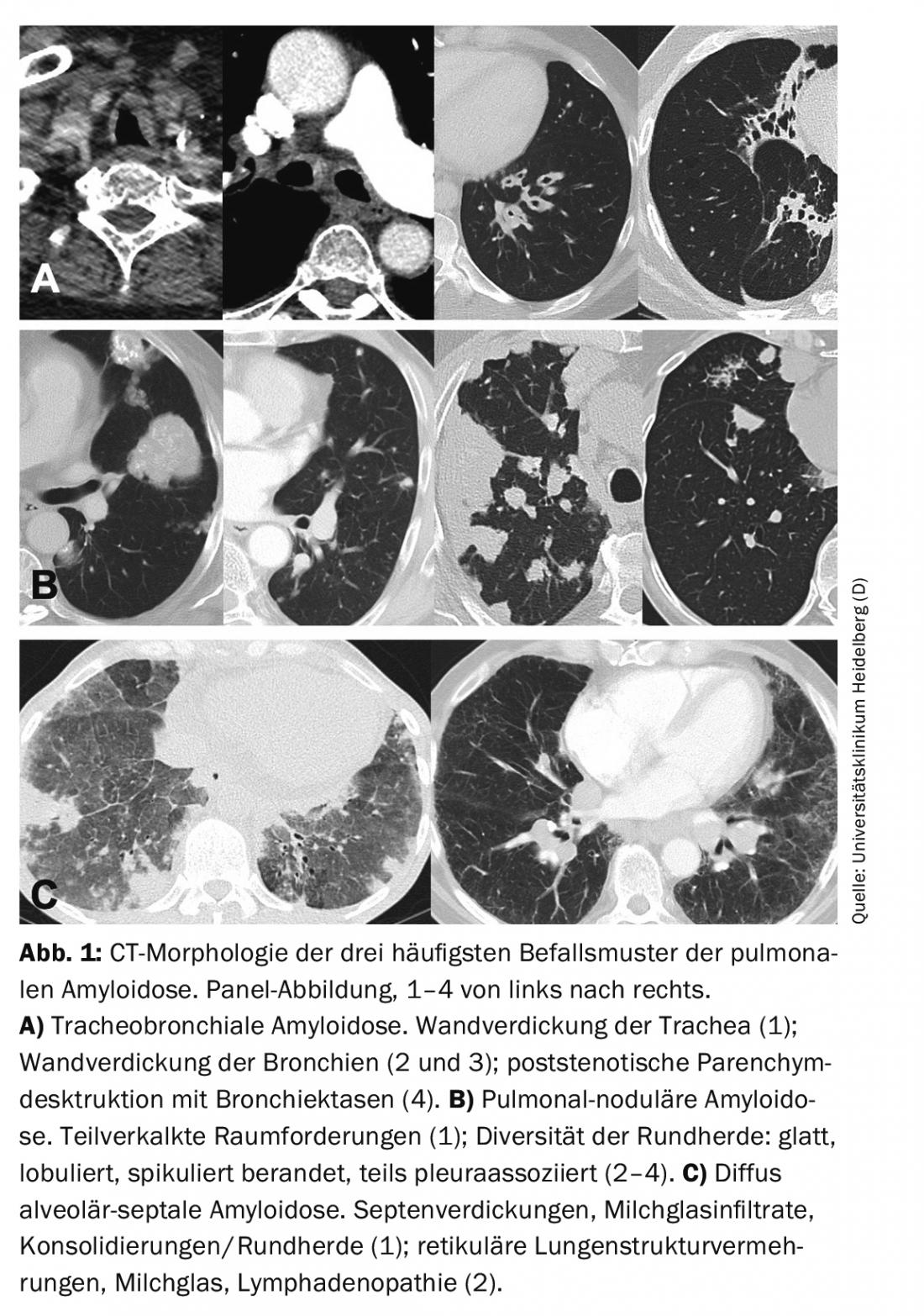
Dependendo do local onde a amiloide é depositada, podem distinguir-se 3 padrões característicos de amiloidose pulmonar: traqueobrônquica (vias aéreas superiores/inferiores), nodular pulmonar e alveolar-septal difusa.
Amiloidose traqueobrônquica
Paciente 1: Vimos uma paciente feminina de 55 anos de idade no diagnóstico em AZ ligeiramente reduzido, IMC 23 kg/m2, com condições pré-existentes de diabetes mellitus tipo 2, hipertensão arterial, abuso persistente de nicotina inalada (35 pack-years) e história de bronquite crónica na infância. Devido à bronquite recorrente, têm sido administradas terapias antibióticas repetidas há mais de um ano. Houve também uma tosse crónica.
Uma atelectasia parcial do lobo médio e 2 focos redondos possíveis foram vistos externamente por raio-X ao tórax. O tórax CT com contraste também mostrou sinais de atelectasia do lobo médio com espessamento muito subtil do brônquio do lobo médio, ainda mais subtil do brônquio superior do lobo direito. Além disso, havia uma linfadenopatia concomitante no lado hilar direito. Até prova em contrário, os resultados foram considerados como uma massa com constrição do brônquio do lobo médio direito e uma suspeita de metástase do gânglio linfático. Foi realizada uma broncoscopia com excisão de amostra, que revelou depósitos amilóides intersticiais e vasculares histologicamente distintos do tipo AL lambda. Não foram detectadas células plasma clonais ou células B na biopsia subsequente da medula óssea. Não foi encontrado amilóide no material de uma biópsia rectal ou na aspiração de tecido adiposo realizada no nosso hospital. A ecocardiografia também não mostrou evidências de envolvimento cardíaco de amiloidose sistémica. No entanto, a electroforese por imunofixação revelou uma gamopatia monoclonal tipo IgG lambda. Fizemos o diagnóstico: Amiloidose brônquica local, tipo AL lambda, com concomitante gamopatia monoclonal tipo IgG lambda.
Após cerca de 1,5 anos, ocorreu uma infecção exacerbada no sentido de uma progressão da amiloidose brônquica com uma oclusão dos brônquios parciais. Realizou-se uma recanalização e dilatação endobrônquica e radioterapia torácica como decisão de caso único com 20 Gy em 10 fracções. Nos meses seguintes, os sintomas melhoraram lentamente. Durante o período de observação posterior de um ano que conhecemos, a condição clínica e radiológica era estável. Não havia provas de progressão da gamopatia monoclonal.
Entre Abril de 2000 e Outubro de 2019, vimos 12 doentes com amiloidose local da nasofaringe, 51 doentes com amiloidose local da laringe e 31 doentes com amiloidose local das vias respiratórias inferiores no Centro de Amiloidose de Heidelberg [6]. Patofisiologicamente, este padrão de envolvimento resulta no aparecimento de placas amilóides submucosas multifocais no epitélio respiratório, causando uma obstrução subtotal total das vias respiratórias dependentes. Dependendo da localização, é possível distinguir um afecto proximal, um médio e um distal [14]. Os pacientes tornam-se frequentemente sintomáticos com tosse, infecções respiratórias recorrentes, hemoptise, estridor inspiratório e rouquidão. A dispneia, frequentemente atribuída erradamente a causas asmáticas, também pode ser a causa, especialmente em casos de estenose proximal grave, por exemplo, estenose subglótica [12].
No diagnóstico da função pulmonar, a amiloidose traqueobrônquica leva à obstrução, especialmente com envolvimento proximal [14]. A morfologia do TAC pode revelar espessamento da traqueia e/ou brônquios (Fig. 1). As estenoses bronquiais levam à distelectasia lobar ou segmentar e podem resultar em pneumonia de retenção e destruição parenquimatosa pulmonar. A calcificação das paredes traqueobrônquicas ocorre com mais frequência. Em contraste com a traqueobronchopatia osteocondroplástica, a parede posterior da traqueia também pode estar envolvida [15]. A broncoscopia revela depósitos esbranquiçados irregulares, que podem subtotalmente restringir totalmente o lúmen. Estas são geralmente difusas e também afectam a parede posterior da traqueia. As lesões são frágeis e propensas a redobrar na biopsia. Histologicamente, o amilóide encontra-se na submucosa e nos vasos sanguíneos, frequentemente associado a plasmócitos e células gigantes. Na subtipagem, AL é normalmente detectável [16].
Na maioria dos casos, a amiloidose traqueobrônquica permanece confinada ao órgão e só raramente é uma expressão da amiloidose AL sistémica [16]. Por conseguinte, a quimioterapia sistémica não é geralmente útil para a doença. Em caso de deficiência grave, por exemplo, devido a bronquite recorrente ou dispneia, são indicadas medidas terapêuticas locais. A ablação endobrônquica do amilóide usando lasers ou fórceps tem provado ser bem sucedida. Em casos excepcionais, também é possível a radiação focalizada, que é potencialmente dirigida contra a doença clonal causal das células B. As infecções pulmonares podem tornar-se fatais devido às estenoses e, portanto, requerem normalmente uma terapia rápida baseada em antibiograma.
Na ausência de hemorragia grave ou infecção pulmonar, o prognóstico da amiloidose traqueobrônquica local é muito bom. Em menos de 20% dos casos, a progressão morfológica clinicamente relevante ou CT da doença ocorre no decurso de 5 anos [6].
Amiloidose nodular pulmonar
Paciente 2: Vimos um paciente de 49 anos em bom estado geral no momento do diagnóstico, IMC de 28 kg/m². Para além de uma condição após abuso de nicotina inalada (15 pack-years, abstinência durante 20 anos), não houve diagnósticos anteriores relevantes.
A apresentação pneumológica inicial foi devida a uma infecção broncopulmonar que durou semanas, febre, suores nocturnos e dispneia ao esforço (estável em termos de peso, sem tosse). A função pulmonar não era notável, sem qualquer restrição de difusão. Oito meses antes da apresentação ao nosso centro, foi realizado um tórax de TC com contraste. Várias compressões parcialmente calcificadas, na sua maioria perifericamente localizadas, eram notórias em ambos os pulmões, que eram novas ou tinham progredido em tamanho em comparação com um exame anterior 3 anos antes. Além disso, várias lesões císticas foram observadas bilateralmente, mas sem massas específicas de malignidade e sem gânglios linfáticos aumentados. Diferentemente, suspeitou-se de micobacteriose atípica. Foi realizada uma broncoscopia flexível, que revelou achados endobrônquicos sem sinais de tumor directo ou indirecto. Na lavagem broncoalveolar do lobo médio, não foi possível detectar nenhum agente patogénico, e não foi possível detectar micobacteriose.
Cinco meses antes da nossa apresentação, foi realizada uma punção guiada por TAC (pneumotórax pós-intervencional sem necessidade de terapia) do lobo inferior direito. Isto mostrou depósitos amilóides intersticiais alterados de forma regressiva AL kappa. Não foi possível detectar células plasma clonais ou células B nem amilóide na biópsia subsequente da medula óssea. Um TAC de seguimento pouco antes da apresentação no nosso centro mostrou constantes granulomas bipulmonares calcificados.
Na apresentação ao nosso centro, realizámos uma aspiração de gordura na qual não foi detectada nenhuma amilóide. A ecocardiografia não mostrou evidências de envolvimento cardíaco de amiloidose sistémica. Por meio de electroforese de imunofixação e medição de cadeias de luz livre no soro, não foi possível excluir uma gamopatia monoclonal de baixo grau do tipo IgG kappa.
Fizemos o diagnóstico: Amiloidose pulmonar local, tipo kappa, com gamopatia monoclonal concomitante V.a. tipo IgG kappa. Um seguimento após 6 meses não mostrou qualquer progressão da gamopatia e ainda não havia provas de amiloidose AL sistémica. Recomendamos um acompanhamento pneumológico regular.
Entre Abril de 2000 e Outubro de 2019, 63 pacientes apresentaram ao Centro de Amiloidose de Heidelberg com amiloidose nodal pulmonar local [6]. Patofisiologicamente, esta manifestação resulta em amiloidomas semelhantes a tumores – depósitos multifocais de amilóide no parênquima pulmonar. Na subtipagem, isto é principalmente AL. Pensa-se actualmente que os linfomas do tecido linfóide associado à mucosa (MALT) são causadores da maioria das amiloidoses nodulares pulmonares [12]. É frequentemente um achado incidental na imagem do tórax. Menos de metade dos doentes em causa apresentam sintomas como tosse, dispneia, hemoptise ou infecções broncopulmonares. Em comparação com a amiloidose traqueobrônquica, o diagnóstico tende portanto a ocorrer mais tarde e a idade média dos doentes com amiloidose nodal pulmonar é mais elevada em comparação com os doentes com amiloidose traqueobrônquica (68 anos vs. 55 anos).
Se os resultados forem pronunciados, é mais provável que se esperem restrições através de diagnósticos da função pulmonar. A morfologia do TC é geralmente dominada por um padrão de focalização nodular com massas solitárias a múltiplas massas e/ou focos redondos. A morfologia dos focos é diversificada com bordas lisas a espiculadas e calcificações opcionais e associação pleural (Fig. 1). A morfologia da TC não pode diferenciar as lesões de doenças malignas ou granulomatosas. Os quistos pulmonares, por vezes com focos redondos associados, e a linfadenopatia também podem fazer parte da variada imagem da TC [17].
Se a neoplasia ou amiloidose sistémica pode ser excluída por diagnóstico diferencial, o prognóstico da amiloidose nodal pulmonar é muito bom. A doença é geralmente branda, com a progressão morfológica do TAC a ocorrer em menos de 36% dos casos ao longo de 5 anos [6]. Os amiloidomas ou quistos funcionalmente limitantes podem ser tratados por excisão cirúrgica com a melhor preservação possível do tecido circundante.
Amiloidose alveolar difusa do septo alveolar
Paciente 3: Vimos uma paciente feminina de 50 anos de idade em AZ reduzido no diagnóstico, IMC 22 kg/m². Cinco meses antes da primeira apresentação no nosso centro, foram administradas terapias antibióticas e anti-obstrutivas para sintomas prolongados de constipação com uma tosse improdutiva, o que não levou a qualquer melhoria dos sintomas. Além disso, a paciente relatou ter notado um aumento do edema do tornozelo e da perna inferior e palpitações cardíacas recorrentes durante cerca de 6 meses. Houve uma restrição severa da capacidade física com dispneia após apenas cerca de meio piso de escadas ou 150 m de caminhada no nível. Até há algumas semanas, tinha perdido uma quantidade considerável de peso (>10% em 6 meses), mas recentemente voltou a engordar com edema clinicamente pronunciado. Ao exame físico, a macroglossia e um inchaço acentuado da base da língua eram evidentes. O paciente teve um historial de picadas recorrentes da língua nos últimos meses. Além disso, eram visíveis hemorragias periorbitárias, que estavam presentes há cerca de um ano. Quando questionada, a paciente declarou que tinha recebido uma cirurgia bilateral da mão para a síndrome do túnel do carpo antes do início dos sintomas pulmonares.
O tórax nativo do CT mostrou opacidades marcadas de vidro de leite bilateralmente com punctum máximo sobre os segmentos do lóbulo inferior. Além disso, houve múltiplas alterações peribronco-vasculares e especialmente alterações inflamatórias subpleurais. O linfoma hilar era suspeito do lado direito. Uma amiloidose alveoloseptal difusa do tipo AL lambda foi detectada por biópsia transbrônquica. A biopsia da medula óssea mostrou uma percentagem de plasmócitos de 80% com restrição da cadeia ligeira lambda. No soro, as cadeias de luz lambda livre foram fortemente elevadas a 7572 mg/l (kappa 8 mg/l).
Em resumo, fizemos o diagnóstico de amiloidose sistémica de AL tipo lambda. Os sintomas iniciais principais incluíam sintomas pulmonares e envolvimento de tecidos moles patognomónicos (macroglossia, hemorragias periorbitárias, secundárias à cirurgia KTS). O estadiamento de órgãos revelou um envolvimento cardíaco de alto grau (NT-proBNP 54 749 ng/l, ecocardiograficamente globalmente ainda bom mas com uma função de bomba significativamente reduzida longitudinalmente com uma espessura septal de 18 mm) e um envolvimento renal (proteinúria 8,2 g/d, eGFR 30 ml/min). Recomendamos uma terapia sistémica relativamente bem tolerada e eficaz com bortezomib e dexametasona. Infelizmente, um mês após a primeira apresentação no nosso centro, ocorreu a exitus lethalis.
O padrão de infestação alveolar-septal difusa está associado ao quadro clínico da doença pulmonar intersticial. Com raras excepções, estes pacientes têm a amiloidose AL sistémica como doença subjacente [12]. Histopatologicamente, são encontrados depósitos amilóides nas paredes dos vasos e septos alveolares, que impedem as trocas gasosas e podem levar à hipertensão arterial pulmonar. O principal sintoma é a dispneia. Os sintomas pulmonares são muitas vezes difíceis de avaliar clinicamente contra o pano de fundo da doença cardíaca geralmente dominante na amiloidose sistémica.
Os diagnósticos da função pulmonar mostram frequentemente restrição e capacidade limitada de difusão de CO O stress leva a uma hipoxemia inadequada. A morfologia da TC mostra geralmente uma proliferação de estruturas pulmonares difusas, tais como espessamento do septo, infiltrados de vidro leitoso e micronódulos (Fig. 1). Além disso, podem ocorrer lesões/consolidações ocupacionais, quistos pulmonares e linfadenopatia [17].
As provas de amiloidose alveolar-septal do pulmão devem provocar um trabalho abrangente para a presença de amiloidose AL sistémica. Raramente, o padrão de envolvimento nodal ou traqueobrônquico pulmonar pode também estar associado à amiloidose sistémica [6,17]. Se houver uma mistura de padrões de infestação, é mesmo muito provável que haja um curso sistémico [16]. A amiloidose sistémica de AL deve, portanto, ser sempre clarificada como um diagnóstico diferencial nos casos de suspeita de amiloidose pulmonar local, especialmente na presença de uma gamopatia monoclonal.
Se nenhuma amiloidose sistémica puder ser detectada, o acompanhamento de perto é de grande importância. Na presença de amiloidose AL sistémica, há uma indicação de quimioterapia sistémica. A escolha do regime de quimioterapia depende principalmente do grau de envolvimento dos órgãos. Por exemplo, a quimioterapia de alta dose com transplante autólogo de células estaminais já não é considerada para pacientes com envolvimento cardíaco significativo ou com uma capacidade de difusão de CO <50%.
No contexto da amiloidose sistémica, ocorrem frequentemente efusões pleurais, geralmente sob a forma de transudado e em até 1/3 dos casos como exsudado [12]. Se as efusões pleurais na amiloidose sistémica forem refratárias à máxima terapia diurética e punções pleurais, o envolvimento pleural deve ser considerado [12].
Diagnóstico diferencial da amiloidose pulmonar local versus amiloidose AL sistémica
A maioria das amiloidoses pulmonares confirmadas histologicamente mostram subtipagem AL. Como descrito acima, a maioria dos casos são amiloidoses locais com um prognóstico muito bom. Contudo, uma vez que qualquer padrão de amiloidose pulmonar também pode ser associado a um curso sistémico de amiloidose, o diagnóstico diferencial de amiloidose AL local versus sistémica é de particular importância, especialmente na amiloidose AL pulmonar recentemente confirmada (Fig. 2) .
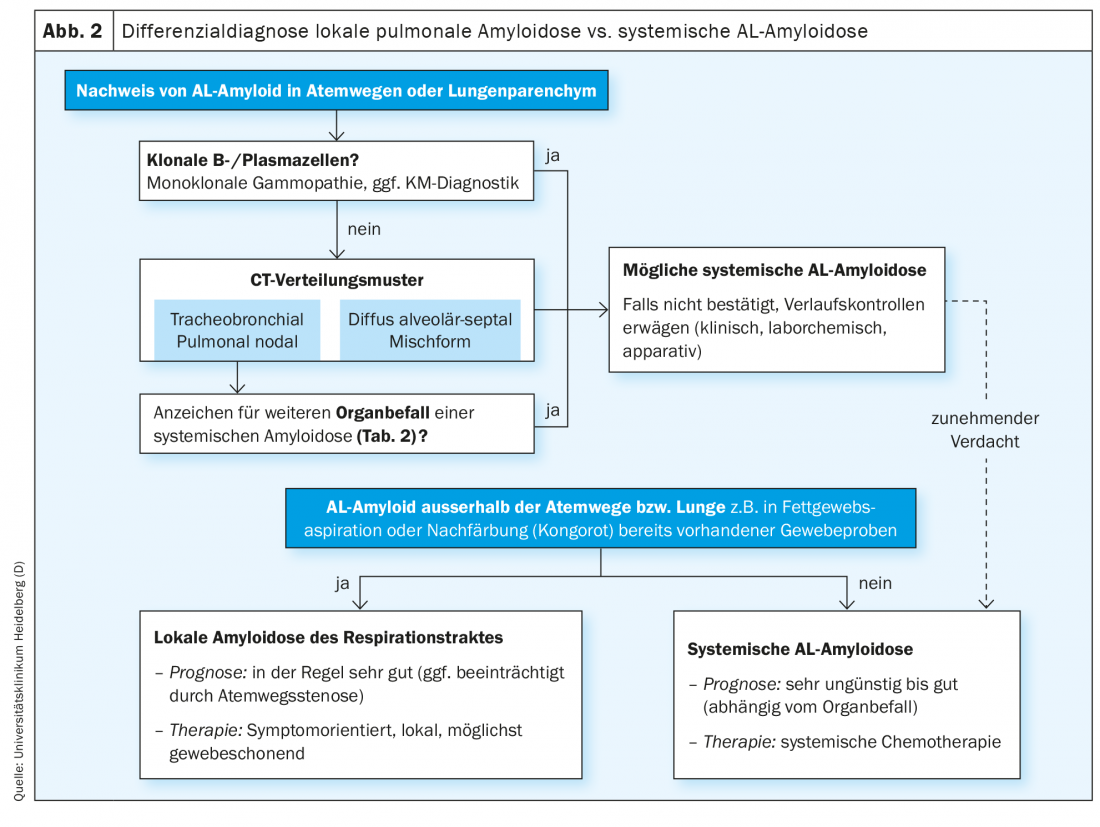
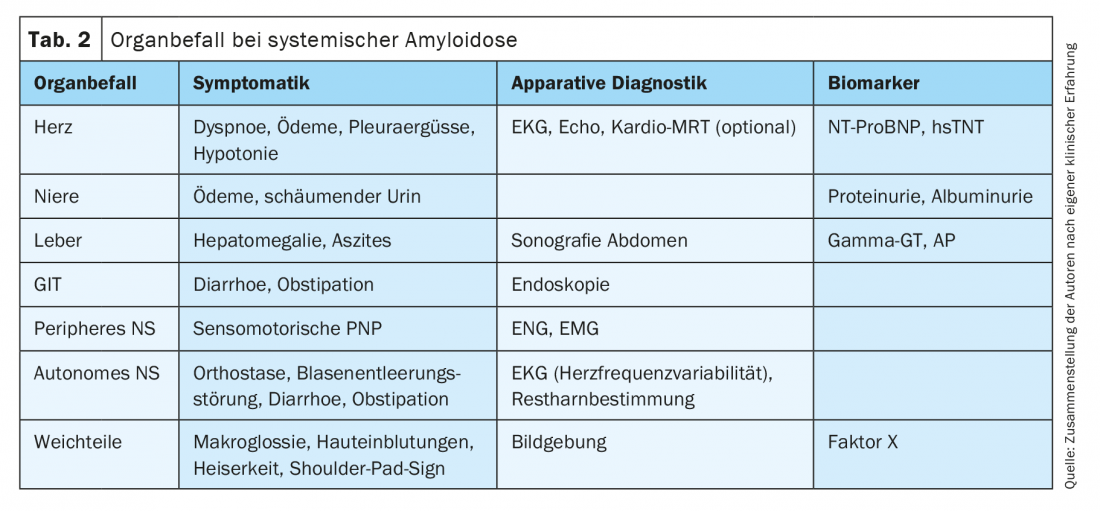
Uma história detalhada e perguntas específicas sobre sintomas concomitantes podem fornecer indicações de outro sistema orgânico afectado no contexto da amiloidose sistémica de AL (Tab. 2) . Síncope, hipotensão arterial e dispneia pulmonar inexplicada ao esforço podem indicar envolvimento cardíaco. A infestação renal manifesta-se geralmente sob a forma de danos glomerulares com síndrome de perda de proteínas; menos frequentemente, a infestação renal-vascular leva directamente à disfunção renal. Para além da urina espumosa, a perda constante de proteínas pode também levar à perda de peso, e no decurso da doença a maioria dos doentes apresenta edema. A infestação do tracto gastrointestinal pode levar a diarreia ou hemorragia. O fígado raramente é clinicamente afectado. No entanto, as perturbações da coagulação também são frequentemente observadas no contexto de uma acentuada afecção dos tecidos moles devido a uma ligação do factor de coagulação X por amilóide. Macroglossia e hemorragias periorbitárias são patognomónicas para amiloidose AL sistémica. A síndrome do túnel do carpo, especialmente de ambos os lados, ou a condição após a cirurgia do KTS é também muito suspeita, pois o retinaculum flexorum do pulso é um local típico e precoce de deposição de proteínas amiloidogénicas [18–20]. Um afecto do sistema nervoso periférico pode manifestar-se em parestesia, termaestésico perturbado e reflexos musculares enfraquecidos, entre outras coisas. A disregulação ortostática grave e a obstipação indicam o envolvimento do sistema nervoso autonómico.
Para além dos sinais vitais, o estadiamento completo dos órgãos instrumentais inclui ECG (baixa voltagem periférica no envolvimento cardíaco, variabilidade limitada da frequência cardíaca na neuropatia autonómica), ecocardiografia (função longitudinal limitada, septo espessado no envolvimento cardíaco) e sonografia hepática. (Tab. 2). Além disso, os biomarcadores de laboratório são os mais importantes indicadores de danos de órgãos no contexto da amiloidose histologicamente comprovada (Tab. 2).
A procura de uma gamopatia monoclonal possivelmente presente é de importância central para o diagnóstico diferencial entre a amiloidose sistémica e a amiloidose AL local. Para assegurar uma sensibilidade suficiente, a electroforese por imunofixação no soro e na urina e a medição de cadeias de luz livre no soro devem ser realizadas em conjunto. Mais de 1/3 dos doentes com amiloidose AL local têm uma gamopatia monoclonal maioritariamente menor, mas apenas cerca de metade destes casos correspondem ao subtipo de depósito AL (kappa ou lambda) [6]. Além disso, cerca de 22% dos doentes com amiloidose AL pulmonar local têm uma doença auto-imune que os acompanha, por exemplo, a síndrome de Sjögren [6]. Estas observações indicam que os doentes com amiloidose AL local têm uma tendência crescente para formar múltiplos clones de células B, embora o contexto fisiopatológico para tal seja ainda desconhecido. Uma gamopatia monoclonal correspondente ao subtipo de depósitos AL pode ser uma expressão do pequeno clone local de células B subjacente à amiloidose local [4,6,11], mas também pode ser uma indicação da presença de amiloidose AL sistémica e deve, portanto, ser normalmente esclarecida através de diagnósticos de medula óssea. No caso de amiloidose AL sistémica, os clones B ou plasmócitos são normalmente detectáveis na medula óssea com restrição de cadeia ligeira correspondente ao subtipo de depósitos AL. Por outro lado, se a gamopatia monoclonal ou a população celular clonal B/plasma não corresponder ao subtipo de deposição AL, não há um risco acrescido de amiloidose AL sistémica.
É mais provável que a amiloidose confinada aos pulmões esteja associada ao abuso da nicotina inalada do que a amiloidose AL sistémica [16,17]. A causa desta associação ainda não é clara, mas poderia possivelmente ser explicada por um aumento da incidência de linfoma causado por agentes nocivos.
Mensagens Take-Home
- A amiloidose pulmonar é uma doença muito rara que pode ser dividida em 3 padrões morfológicos principais da TC: traqueobrônquica, nodal pulmonar e intersticial ou alveolar-septal.
- Os doentes afectados são frequentemente assintomáticos (especialmente os nódulos pulmonares) e a infestação clinicamente sintomática pode normalmente ser bem controlada com medidas locais.
- Na amiloidose pulmonar local (mais comum), o prognóstico a longo prazo é normalmente muito bom, mas as progressões locais são comuns.
- No entanto, qualquer padrão de amiloidose pulmonar pode também ser uma expressão de amiloidose AL sistémica com um prognóstico potencialmente muito desfavorável.
- Especialmente na presença de uma gamopatia monoclonal, de um padrão alveolar-septal de infestação ou de sintomas indicando uma infestação sistémica, uma amiloidose AL sistémica deve ser esclarecida como um diagnóstico diferencial.
Literatura:
- Merlini G, Bellotti V: Mecanismos moleculares da amiloidose. N. Engl. J. Med 2003; 349(6): 583-596.
- Benson MD, Buxbaum JN, Eisenberg DS, et al: Amyloid nomenclature 2018: recommendations by the International Society of Amyloidosis (ISA) nomenclature committee. Amyloid 2018; 25(4): 215-219.
- Kyle RA, Linos A, Beard CM, et al: Incidence and natural history of primary systemic amyloidosis in Olmsted County, Minnesota, 1950 to 1989. Blood 1992; 79(7): 1817-1822.
- Stuhlmann-Laeisz C, Schönland SO, Hegenbart U, et al: AL amiloidose com uma neoplasia localizada de células B. Arco de Virchows 2019; 474(3): 353-363.
- Meijer JM, Schonland SO, Palladini G, et al: síndrome de Sjögren e amiloidose cutânea nodular localizada: coincidência ou uma entidade clínica distinta? Arthritis Rheum 2008; 58(7): 1992-1999.
- Basset M, Hummedah K, Kimmich C, et al: amiloidose de cadeia ligeira de imunoglobulina localizada: novos conhecimentos, incluindo factores prognósticos para a progressão local. Am J Hematol 2020; 1158-1169.
- Veelken K, Hegenbart U, Schönland SO, Blank N.: Amiloidose local e sistémica de cadeia ligeira em doentes com doenças reumáticas. Z Rheumatol 2020; 660-668.
- Dittrich T, Benner A, Kimmich C, et al.: Análise de desempenho de sistemas de estadiamento de biomarcadores cardíacos de amiloidose AL com especial enfoque na insuficiência renal e arritmia atrial. Haematologica 2019; 104(7): 1451-1459.
- Dittrich T, Bochtler T, Kimmich C, et al.: Os pacientes com amiloidose AL com baixos níveis de cadeia de luz livre de amiloidogénio no primeiro diagnóstico têm um prognóstico excelente. Sangue 2017; 130(5): 632-642.
- Kimmich C, Schönland S, Kräker S, et al: Amilóide em manchas de medula óssea em amiloidose sistémica de cadeia de luz. Amyloid 2017; 24(1): 52-59.
- Baumgart J-V, Stuhlmann-Laeisz C, Hegenbart U, et al: Local vs. amiloidose pulmonar sistémica – impacto no diagnóstico e gestão clínica. Arco de Virchows 2018; 473(5): 627-637.
- Milani P, Basset M, Russo F, et al: O pulmão na amiloidose. Eur Respir Rev 2017; 26(145): 170046.
- Ussavarungsi K, Yi ES, Maleszewski JJ, et al: Relevância clínica da amiloidose pulmonar: uma análise de 76 casos derivados de autópsia. Eur Respir J 2017; 49(2): 1602313.
- O’Regan A, Fenlon HM, Beamis JF, et al: Amiloidose traqueobrônquica. A experiência da Universidade de Boston de 1984 a 1999. Medicina (Baltimore) 2000; 79(2): 69-79.
- Czeyda-Pommersheim F, Hwang M, Chen SS, et al: Amiloidose: Modern Cross-sectional Imaging. Radiografias 2015; 35(5): 1381-1392.
- Rech JS, Arnulf B, de Margerie-Mellon C, et al: Amiloidose do tracto respiratório inferior: apresentação, sobrevivência e factores de prognóstico. Uma série de casos consecutivos multicêntricos. Am J Hematol 2019; 94(11): 1214-1226.
- Brandelik SC, Heussel CP, Kauczor HU, et al: Características da TC na amiloidose do sistema respiratório – Análise exaustiva numa coorte de centro de referência terciária. Eur J Radiol 2020; 129: 109123.
- Kelly JJ: Neuropatias periféricas associadas a proteínas monoclonais: Uma revisão clínica. Muscle & Nerve 1985; 8(2): 138-150.
- Haan J, Peters WG: doença amilóide e do sistema nervoso periférico. Neurologia e Neurocirurgia Clínica 1994; 96(1): 1-9.
- Sperry BW, Reyes BA, Ikram A, et al: Tenosynovial and Cardiac Amyloidosis in Patients Undergoing Carpal Tunnel Release. J Am Coll Cardiol 2018; 72(17): 2040-2050.
InFo PNEUMOLOGIA & ALERGOLOGIA 2020; 2(4): 11-17