A doença sistémica auto-imune APS (síndrome antifosfolipídica) caracteriza-se principalmente por manifestações trombóticas. Como uma trombofilia adquirida, é indicada a tromboprofilaxia variável de drogas.
A síndrome antifosfolipídica (SAF) é uma doença sistémica auto-imune caracterizada pela ocorrência de trombose venosa e/ou arterial e/ou complicações na gravidez [1]. A patogénese da APS baseia-se na presença de anticorpos dirigidos contra uma variedade de complexos fosfolípidos/proteicos [2]. A descoberta e o conhecimento destes componentes abrange várias décadas (Tab. 1).

O patomecanismo de como os anticorpos antifosfolípidos (aPL) levam à activação da coagulação é multifactorial (Tab. 2). Existem abordagens diferentes dos estudos humanos e animais que provam o envolvimento de aPL [2]. A EPA pode ocorrer isoladamente (EPA primária) ou no contexto de outras doenças auto-imunes (por exemplo, LES, doenças malignas). Uma forma rara mas muito grave de APS é a síndrome catastrófica dos antifosfolípidos com o envolvimento simultâneo de três ou mais sistemas de órgãos [3].

Anticorpos antifosfolípidos (aPL)
Os anticorpos antifosfolípidos são um grupo heterogéneo de anticorpos adquiridos que são dirigidos contra complexos fosfolípidos-proteínas e mostram uma grande afinidade por superfícies carregadas negativamente. Os antigénios alvo mais importantes (proteínas) incluem β2-glycoprotein-I (β2-GPI) e protrombina. Outros antigénios como a proteína C activada, proteína S, anexina V, LDL oxidado e factor XII também foram descritos (Fig. 1) [2,4]. As interacções da aPL em superfícies fosfolipídicas carregadas negativamente causam várias reacções tais como inibição da regulação hemostática (por inibição da proteína C ou antitrombina, ou deslocamento da anexina V dos trofoblastos na placenta), activação das plaquetas (por estimulação do tromboxano A2), up-regulation das moléculas de adesão e do factor de tecido nas células endoteliais, e activação do complemento. (Tab.2).
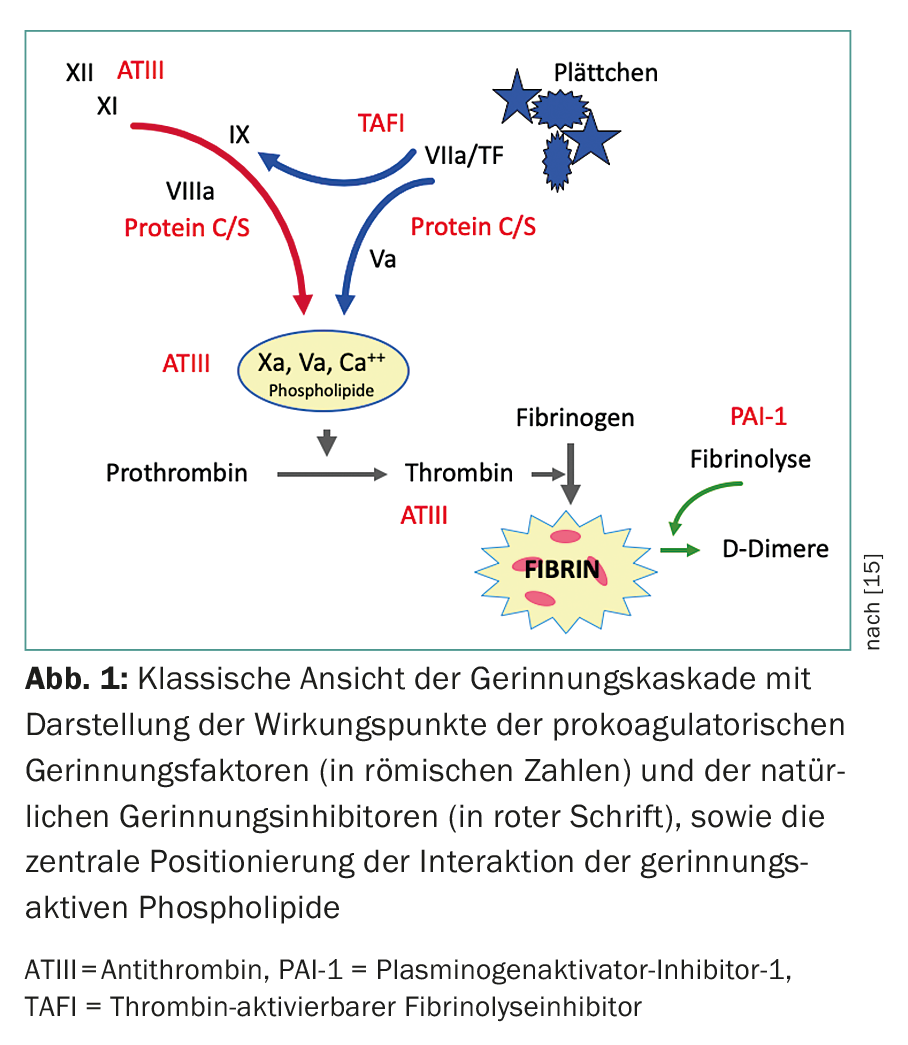
aPL pode levar a um prolongamento do tempo de coagulação nos testes de coagulação dependentes de fosfolípidos (por exemplo Quick/INR ou APTT), uma vez que interferem ou bloqueiam os fosfolípidos do complexo protrombinase necessário para o curso da cascata de coagulação. Este fenómeno in vitro levou ao nome enganador “lúpus anticoagulante” em 1952, quando este efeito “anticoagulante” foi observado em doentes com lúpus eritematoso em testes de laboratório.
Critérios de classificação
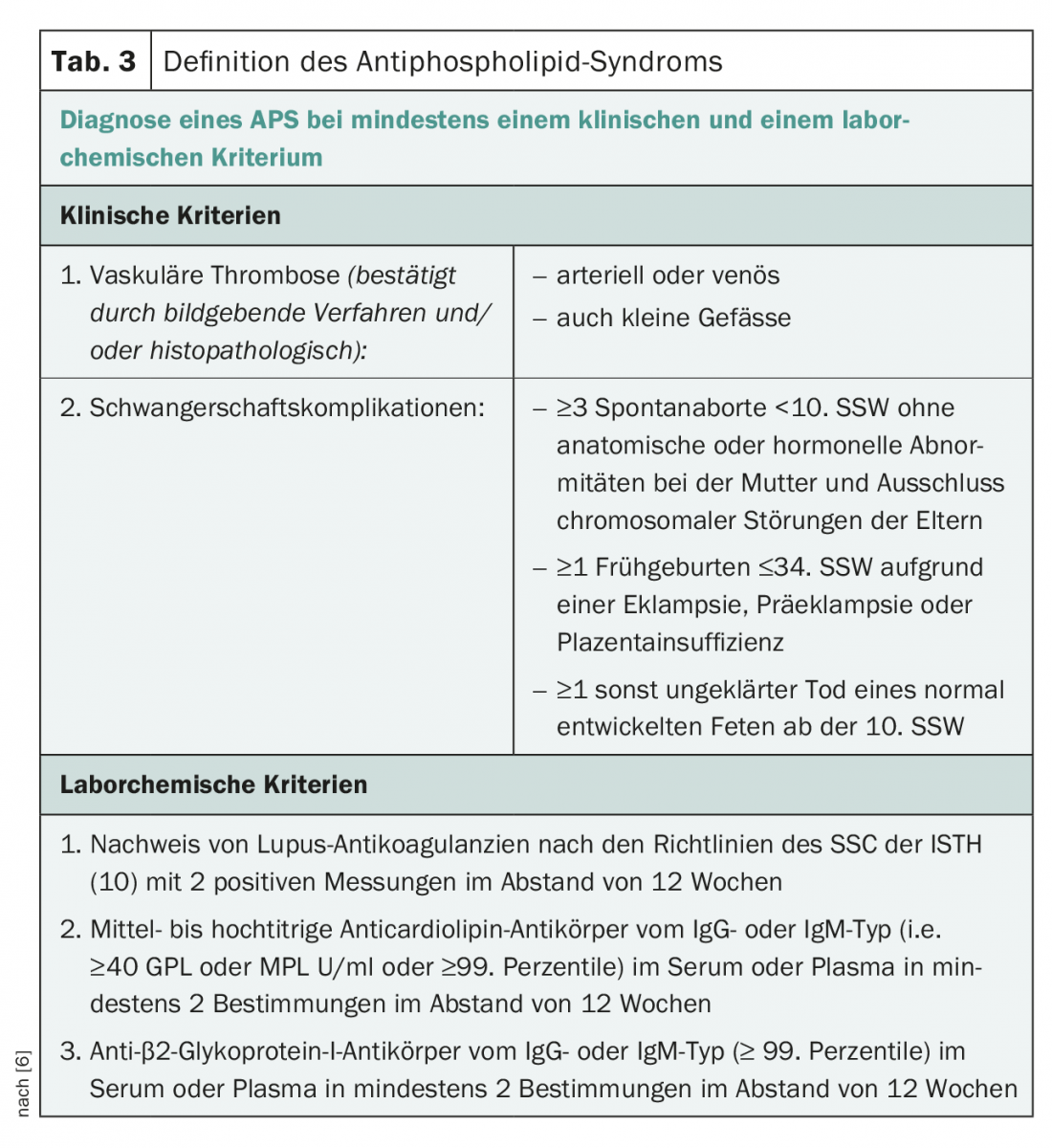
Os critérios de classificação do SPG foram formulados pela primeira vez durante um workshop em Sapporo (Japão) em 1998 e revistos em 2005 no “11th International Congress on Antiphospholipid Antibodies” em Sydney [5,6]. Assim, é necessário um critério clínico (trombose venosa e/ou arterial na micro ou macrocirculação e complicações obstétricas) mais um critério laboratorial (dois positivos no prazo de 12 semanas para o anticoagulante lúpus, anticorpo anticardiolipina ou anticorpo anti-COPY4) para definir a APS definitiva (tab. 3).
Diagnóstico do SPG
Para a detecção de aPL (LA, aCL-IgGG /-IgM, β2GPI-IgGG/-IgM), vários métodos de teste são combinados de acordo com as recomendações internacionais [7,8]. Basicamente, os anticoagulantes lúpus anticoagulantes são identificados funcionalmente através de testes de coagulação. aCL e β2GPI anticorpos, por outro lado, são detectados imunologicamente, por ELISA ou por ensaio de quimioluminescência. A determinação de aPL duas vezes em intervalos de pelo menos doze semanas é necessária para evitar a detecção de quaisquer anticorpos transitórios que possam ocorrer, por exemplo, no contexto de infecções. A figura 2 ilustra o algoritmo de diagnóstico, que se destina a ilustrar a complexidade das etapas de diagnóstico em casos de suspeita de APS.
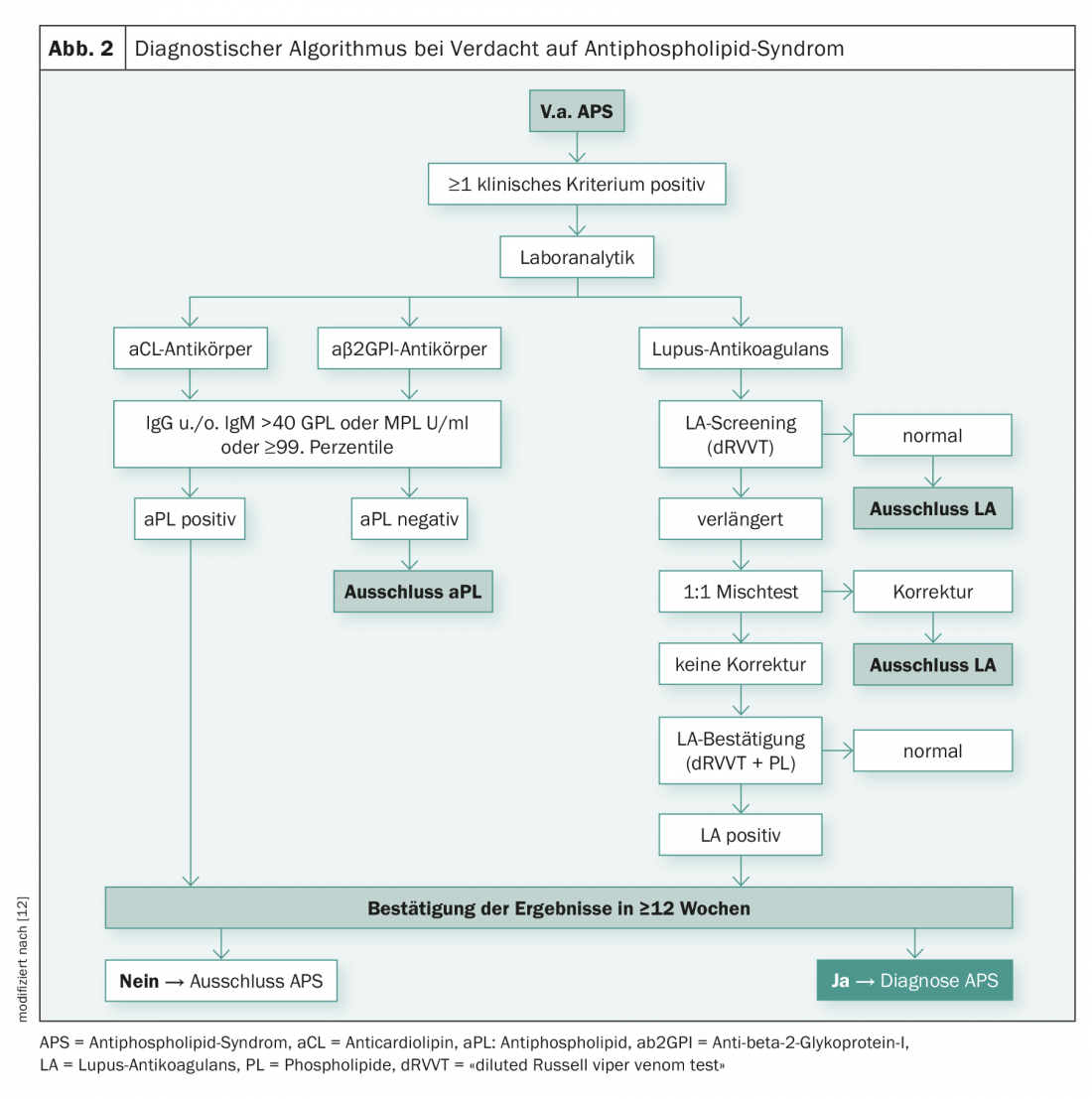
Testes de coagulação – Lúpus anticoagulante: A Sociedade Internacional de Trombose e Hemostasia (ISTH) define uma estratégia em três fases em dois procedimentos de testes LA diferentes cada um [7]:
- Teste de rastreio: prolongamento de um tempo de coagulação dependente de fosfolípidos (>percentil 99);
- Teste misto: Confirmação de um inibidor de acção imediata e exclusão de uma deficiência do factor de coagulação;
- Teste de confirmação: Confirmação de que o inibidor é dependente de fosfolípidos. O terceiro passo é necessário para assegurar que o inibidor não seja dirigido contra um factor de coagulação específico.
Testes imunológicos – aCL/aβ2GPI-ELISA: Além dos testes de coagulação, os anticorpos aPL (aCL-IgG e -IgM assim como aβ2GPI-IgG e -IgM) são determinados imunologicamente pelo ELISA. Tendo em conta as propriedades variáveis dos antigénios e a falta de materiais de referência, a padronização dos sistemas de teste é muito difícil. Os títulos de anticorpos IgG e IgM são expressos em unidades GPL e MPL normalizadas internacionalmente. Os resultados positivos são definidos como títulos ≥40 unidades GPL/MPL (U/ml) ou ≥99th percentil do respectivo método [8].
Embora os testes anti-β2GPI tenham uma maior especificidade para a trombogenicidade do que o aCL, também não estão ainda suficientemente normalizados. Os métodos de teste que utilizam o domínio recombinante 1 de β2GPI, ou seja, o epitopo para a ligação aPL, como antigénio, são considerados promissores [9,10]. Trabalhos recentes mostraram que os anticorpos dirigidos contra este epitopo estão mais fortemente associados a sintomas clínicos. Contudo, são necessários mais esforços para conseguir uma melhor normalização dos diferentes métodos de ensaio, bem como uma melhor caracterização da APL clinicamente relevante.
Clínica
APS é uma doença multiorgânica com múltiplas manifestações, que é classificada como uma trombofilia adquirida. O quadro clínico é caracterizado por tromboses arteriais e/ou venosas recorrentes de vasos pequenos ou grandes com localização típica ou atípica, bem como complicações vasculares da gravidez. Além disso, foram descritas outras manifestações clínicas, tais como disfunção neurológica (epilepsia, demência), sintomas dermatológicos (livedo reticularis/racemosa, necrose acral), doença cardíaca valvular (endocardite trombótica não bacteriana) e enfarte do miocárdio, doenças renais (trombose da artéria renal/venosa renal, microangiopatia trombótica), doenças oculares (amaurose fugax, oclusões de vasos retinais e/ou coróides) e trombocitopénias. Nem sempre é claro nestes quadros clínicos se o aPL representa um epifenoma ou se está patogeneticamente envolvido.
Indicação: o rastreio aPL deve ser iniciado se houver uma elevada probabilidade da presença de APS com base num critério clínico (tab. 3) . Não é recomendado o rastreio não direccionado do aPL em indivíduos assintomáticos, dada a baixa especificidade dos sistemas de teste.
O diagnóstico de APS baseia-se na detecção repetida de LA, aCL ou aβ2GPI a intervalos de 12 semanas, como mencionado acima. O perfil aPL (isótipo e número de testes positivos) também pode ser utilizado para avaliar o risco e a fiabilidade dos resultados [11]. Foi demonstrado que 98% dos pacientes triplamente positivos, 84% dos duplamente positivos e 40% dos positivos simples ainda eram positivos após doze semanas. Estes dados sublinham que o triplo perfil positivo é um resultado robusto de laboratório. Além disso, um perfil triplo positivo corresponde a uma classificação para APS de alto risco, enquanto um perfil positivo duplo ou único corresponde a um risco moderado ou baixo para APS, respectivamente.
Terapia
A estratégia de tratamento quando a APL é detectada depende principalmente de os pacientes estarem assintomáticos com apenas resultados laboratoriais positivos ou pacientes sintomáticos com complicações manifestas. Para pacientes assintomáticos, aplica-se a mesma recomendação de tromboprofilaxia que para outros pacientes trombofílicos. No entanto, no caso de APS manifesta com sintomas correspondentes, deve ser feita uma distinção quanto à presença ou não da forma primária ou secundária. No caso da EPA primária sem evidência de outra doença subjacente, a terapia tem como único objectivo prevenir mais complicações de trombose. No caso da SAF secundária, o primeiro passo é tratar a doença subjacente ou minimizar os factores de risco típicos de complicações cardiovasculares, em paralelo com o tratamento da síndrome trombótica.
Anticoagulação (ASA, VKA, NOAK): Em pacientes com APS com trombose venosa, recomenda-se a terapia inicial com heparina não fracionada (UFH) ou de baixo peso molecular (NMH), seguida de terapia a longo prazo com antagonistas de vitamina K (VKA). Devido ao elevado risco de trombose recorrente (dependendo, entre outras coisas, se o paciente pertence ao grupo de alto risco com resultados triplamente positivos da APL) após a interrupção da anticoagulação oral, a anticoagulação oral a longo prazo com INR 2-3 alvo é a terapia de escolha na maioria dos casos, pelo menos enquanto os resultados laboratoriais permanecerem positivos [12]. Isto reduz significativamente as recidivas tromboembólicas. No entanto, revela-se insuficiente em alguns casos. Nesses casos, pode ser considerada a terapia a longo prazo com NMH ou o aumento da INR alvo para 3-4.
Os pacientes com trombose arterial ou pacientes com SPAs triplo positivo que estão em alto risco de eventos tromboembólicos beneficiam mais da anticoagulação sistémica do que de agentes antiplaquetários. A anticoagulação ao longo da vida é indicada em tais pacientes.
A utilização dos novos anticoagulantes orais directos (NOAKs) é controversa [12]. Os NOAK não requerem controlos laboratoriais e foram considerados pelo menos tão eficazes e seguros como VKA ou NMH no tratamento ou prevenção do tromboembolismo venoso e arterial. No entanto, estudos prospectivos e retrospectivos demonstraram que os NOAK são menos eficazes do que a warfarina em doentes com SAF de alto risco [13,14]. A actual base de evidência para a utilização e eficácia dos NOAK em doentes com SAF é ainda insuficiente e são necessários estudos prospectivos adicionais para avaliar o seu valor na SAF.
A anticoagulação em mulheres com complicações recorrentes na gravidez devido à SAF tem como objectivo preservar o feto ou aumentar a taxa de nascimento vivo. Para o tratamento de mulheres grávidas, recomenda-se uma combinação de NMH em dose subterapêutica (1×100 E/kgKG/dia) e aspirina (1×100 mg/dia). A anticoagulação deve ser iniciada logo que possível após a confirmação da gravidez e deve ser continuada até seis semanas pós-parto.
Imunossupressão: O tratamento com imunossupressores além da anticoagulação é indicado principalmente na presença de APS secundária, trombocitopenia ou síndrome antifosfolipídica catastrófica (CAPS). A utilização de imunossupressores na SAF para a profilaxia de outros eventos tromboembólicos está também a ser discutida, uma vez que a imunossupressão tem uma influência favorável em títulos elevados de APL e poderia assim resultar possivelmente numa incidência reduzida de oclusão vascular. No entanto, não estão disponíveis recomendações concretas sobre o tipo e duração da imunossupressão. Estes são derivados da experiência com o tratamento de trombocitopenia auto-imune ou inibidores do factor de coagulação (por exemplo, terapia única ou combinada com prednisona/ciclopfosfamida/rituximab).
Mensagens Take-Home
- A síndrome dos antifosfolípidos (SAF) é uma doença auto-imune sistémica com um evento patogénico primário: formação de auto-anticorpos contra complexos fosfolípidos/proteínas coagulantes.
- Apesar do fenómeno paradoxal do prolongamento dos testes de coagulação global in vitro, o quadro clínico do paciente com SAF não se caracteriza por hemorragia mas sim por manifestações trombóticas.
- O diagnóstico de APS requer resultados laboratoriais (lúpus anticoagulante e/ou anticorpos antifosfolípidos de médio-alto positivo) combinados com trombose clínica na micro ou macrocirculação.
- A APS é considerada uma trombofilia adquirida e necessita de tromboprofilaxia com drogas variáveis como terapia, adaptada à intensidade e duração, dependendo do envolvimento dos órgãos e da extensão das manifestações trombóticas. As heparinas ou antagonistas de vitamina K continuam a ser anticoagulantes de primeira escolha.
Literatura:
- Cervera R, Espinosa, Khamashta MA: Síndrome dos antifosfolípidos nas doenças auto-imunes sistémicas, 2ª edição. Elsevier, Amsterdão, 2016.
- Giannakopoulos B, Krilis SA: A patogénese da síndrome dos antifosfolípidos. N Engl J Med 2013; 368: 1033-1044.
- Asherson RA, et al: Catastrophic antiphospholipid syndrome: declaração de consenso internacional sobre critérios de classificação e directrizes de tratamento. Lupus 2003; 12: 530-534.
- Arnout J, Vermylen J: Current status and implications of autoimmune antiphospholipid antibodies in relation to thrombotic disease; J Thromb Heamost 2003; 1: 931-942.
- Wilson WA, et al: Declaração de consenso internacional sobre os critérios preliminares de classificação da síndrome antifosfolipídica definitiva: relatório de um workshop internacional. Arthritis Rheum 1999; 42: 1309-1311.
- Miyakis S, et al: Declaração de consenso internacional sobre uma actualização dos critérios de classificação para a síndrome dos antifosfolípidos (APS) definitiva. J Thromb Haemost 2006; 4: 295-306.
- Pengo V, et al: Actualização das directrizes para a detecção de lúpus anticoagulante. Subcomité sobre lúpus anticoagulante/antifosfolípido do comité científico e de normalização da sociedade internacional sobre trombose e hemostasia, J Thromb Haemost 2009; 7: 1737-1740.
- Devreese KMJ, et al:. Subcomissão sobre Lúpus Anticoagulante/Antifosfolipídico Anticorpos, Critérios laboratoriais para a síndrome antifosfolipídica: comunicação do SSC do ISTH, J Thromb Haemost 2018; 16: 809-813.
- De Craemer AS, Musial J, Devreese KM: Papel dos anticorpos anti-domínio 1-beta2 da glicoproteína I no diagnóstico e estratificação do risco da síndrome antifosfolipídica. J Thromb Haemost 2016; 14: 1779-1787.
- Pengo V, et al: Síndrome dos antifosfolípidos: anticorpos para o domínio 1 da beta2-glicoproteína 1 classificam correctamente os doentes em risco. J Thromb Haemost 2015; 13:782-7.
- Pengo V, et al: A confirmação da positividade inicial dos anticorpos antifosfolípidos depende do perfil dos anticorpos antifosfolípidos. J Thromb Haemost 2013; 11: 1-5.
- Garcia D, Erkan D: Diagnóstico e Gestão da Síndrome Antifosfolipídica. N Engl J Med 2018; 378: 2010-2021.
- Martinelli I, et al: Trombose recorrente em doentes com anticorpos antifosfolípidos tratados com antagonistas de vitamina K ou rivaroxaban. Haematologica 2018;103(7): 315-317.
- Pengo V, et al: Rivaroxaban vs warfarin em doentes de alto risco com síndrome antifosfolipídica. Sangue 2018; 132(13): 1365-1371.
- Tsakiris D, Bachofner A: Coagulação intravascular disseminada no doente com tumor. Info@Oncologia 2017; 7:19-21.
InFo ONcOLOGIA & HaEMATOLOGIA 2019; 7(1): 22-25.