A eosinofilia no sangue é definida por um número >500 eosinófilos por μl. Se um número de >1500/μl for medido pelo menos duas vezes no espaço de duas semanas, está-se perante uma hipereosinofilia (HE). A síndrome hipereosinofílica (HES) é quando à HE se juntam lesões orgânicas. A forma de classificar melhor a HES e de diagnosticar a EGPA, por exemplo, foi debatida no Congresso DGIM 2023.
Desde 2012, a HE e a HES foram subdivididas com base em diferentes etiologias em formas primárias e formas reactivas, bem como em HE e HES idiopáticas, nas quais não é detectada uma doença subjacente clonal ou reactiva. Devido ao número crescente de marcadores e alvos, foi publicada uma atualização desta classificação no início deste ano [1]. Entre outras coisas, a atualização estipulou que uma contagem medida de >1500/µl em pelo menos duas ocasiões, com um intervalo de pelo menos duas semanas, é necessária para definir a EH – na versão anterior, o intervalo de tempo era ainda de quatro semanas.
Dr. Peter Valent, do Hospital Universitário AKH de Viena, e colegas escrevem que, para além da presença de HE no sangue e/ou nos tecidos e da lesão de órgãos associada à HE como terceiro critério, a HES é definida pela exclusão de outra doença ou patologia subjacente como causa primária da lesão de órgãos.
Os autores sublinham que a HES não é um diagnóstico definitivo nem uma doença imunológica ou hematológica definida. Em vez disso, a etiologia contribuinte deve ser identificada em todos os doentes com EES, se possível. Se não for identificada qualquer doença subjacente, o diagnóstico final é de EH idiopática (EHIS).
Os subtipos de EES são doenças mieloproliferativas (14%) e linfoproliferativas (14%), idiopáticas (42%), associadas (9%) e sobrepostas (21%). O grupo de doenças associadas também inclui a granulomatose eosinofílica com poliangiite (EGPA), anteriormente conhecida como síndrome de Churg-Strauss.
Os anticorpos citoplasmáticos anti-neutrófilos (ANCA) são detectados em cerca de 40% dos doentes com EGPA. Dada a semelhança das manifestações clínicas da EGPA (e de outras síndromes relacionadas com a HE) com as características clássicas da HES, Valent et al. concluíram que estas síndromes devem ser classificadas como EES se os critérios de EES forem cumpridos. Se não for esse o caso, o diagnóstico final deve ser a síndrome correspondente e não a HES.
| Critérios de classificação ACR para vasculite Eosinofilia >10% (opcional) Eosinofilia tecidular Asma (>90%) Anomalias do NNH (aprox. 50%) Infiltrados pulmonares voláteis (50-70%) Neuropatia (aprox. 75%) |
| de acordo com [3] |
A deteção da vasculite não é frequentemente possível
“Para distinguir entre um HES e um EGPA, é preciso saber que o EGPA precisa de evidência de vasculite”, explicou o Dr. Christof Iking-Konert, chefe do departamento de reumatologia do Stadtspital Triemli em Zurique [2]. Referiu-se a este facto. De acordo com os critérios de classificação do Colégio Americano de Reumatologia (ACR) [3] (caixa).
A EGPA é a forma mais rara de vasculite associada a ANCA, com uma prevalência de 24/1 milhão. O primeiro desafio de diagnóstico é, portanto, pensar nela primeiro. O segundo obstáculo: A evidência necessária de vasculite na histologia é muitas vezes difícil porque, por exemplo, uma biopsia não é viável em doentes muito doentes. Normalmente, a EGPA é apenas suspeita, pelo que são utilizados substitutos da vasculite, como explicou o Dr. Iking-Konert. Segundo ele, se um ou mais destes factores estiverem presentes, pode presumir-se uma correlação com a vasculite:
- Hemorragia alveolar
- Púrpura palpável
- Enfarte do miocárdio devido a coronariite comprovada
- Hematúria associada a contagens de glóbulos vermelhos ou >10% de glóbulos vermelhos dismórficos ou hematúria e 2+ proteinúria.
- Mononeurite ou mononeurite múltipla
- MPO ANCA e qualquer tipo de manifestação sistémica.
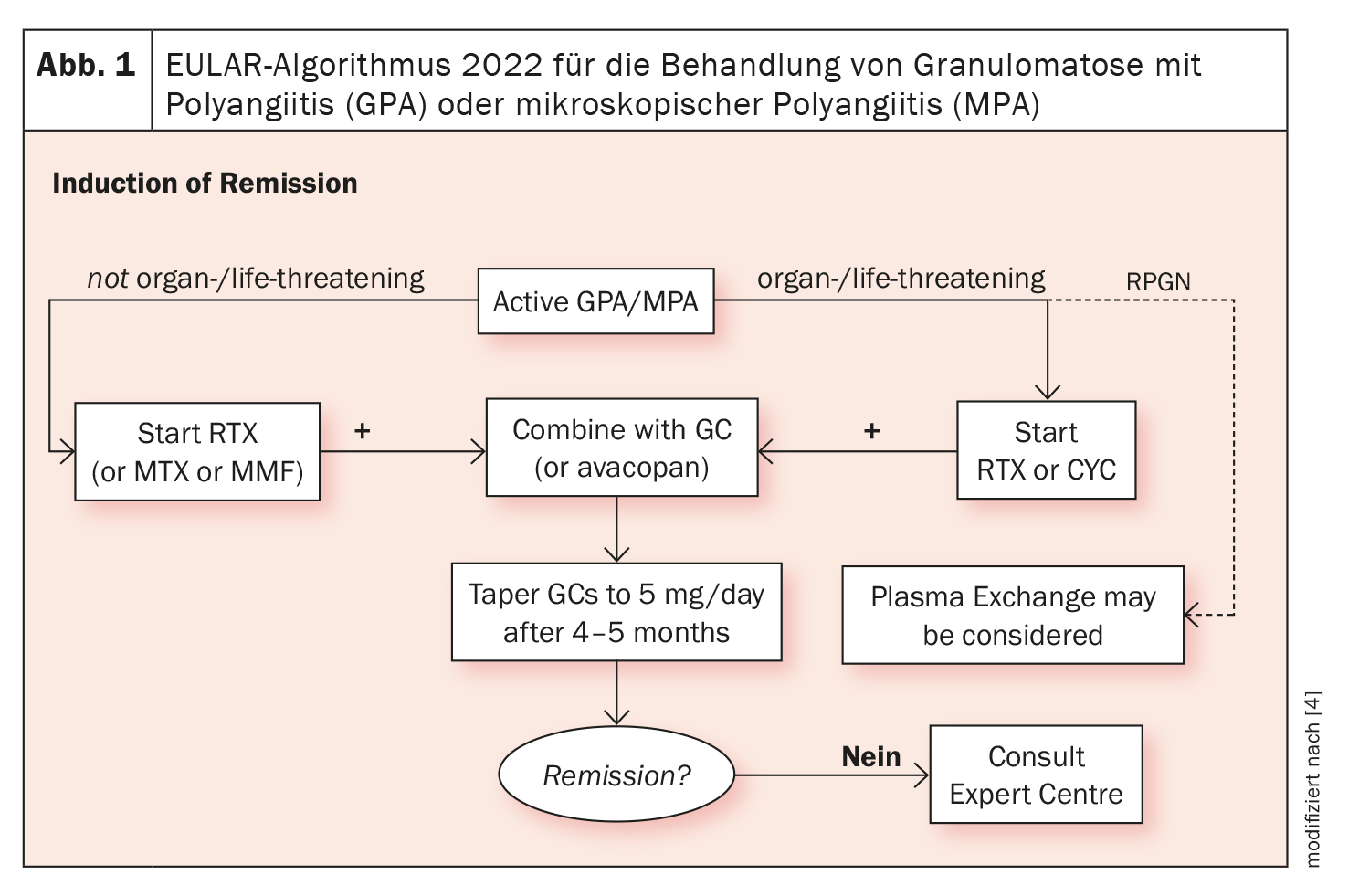
Os produtos biológicos estão disponíveis para o tratamento da EGPA há mais de uma década. Para além do rituximab, foram estabelecidas terapias direccionadas com anticorpos contra a IL-5. A Dra. Iking-Konert referiu-se à diretriz da EULAR sobre a gestão da vasculite associada a ANCA, que será actualizada em 2022: Esta recomenda o início de uma terapia com ciclofosfamida (CYC) ou rituximab (RTX), que são combinados com cortisona, no caso de EGPA com risco para os órgãos ou para a vida. Em doenças não orgânicas ou potencialmente fatais, o bloqueio da IL-5 está indicado para cursos refractários ou graves (Fig. 1) [4].
Congresso: DGIM Industriesymposium GSK
Literatura:
- Valent P, et al.: Allergy 2023; 78: 47–59.
- Industrie Symposium «Und täglich grüsst das Immunsystem: seltene Autoimmunerkrankungen erkennen und behandeln». 129. Kongress der DGIM, 23.04.2023; Veranstalter: GSK.
- Jennette JC, et al.: Arthritis Rheum 1994; 37: 187–192.
- Hellmich B, et al.: Ann Rheum Dis 2023; doi: 10.1136/ard-2022-223764.
| Imagem da capa: Micrografia de grande ampliação de vasculite eosinofílica consistente com a síndrome de Churg-Strauss, abreviada CSS. H&E stain. Fonte: Nephron, wikimedia |
InFo RHEUMATOLOGIE 2023; 5(1): 17