A atrofia muscular espinhal é uma doença genética rara caracterizada pela perda de neurónios motores na medula espinhal e no tronco cerebral inferior. Entretanto, os peritos concordam que a gestão terapêutica deve ser orientada para as necessidades individuais e objectivos subjectivos da pessoa em causa, especialmente com o aumento da idade. Porque estes têm uma grande influência sobre a qualidade de vida.
Um em cerca de 10.000 recém-nascidos tem um defeito genético que perturba a transmissão de impulso dos neurónios motores. A atrofia muscular espinal (AME) é uma doença muscular recessiva rara, predominantemente autossómica, na qual as células motoras do corno anterior e os núcleos nervosos motores cranianos degeneram. É a doença hereditária mais comum que resulta na morte na infância e é frequentemente diagnosticada numa idade muito precoce – embora não exclusivamente [1,2]. A SMA também pode não se tornar aparente até à idade adulta [3]. Esta heterogeneidade requer portanto uma terapia individual que tenha em conta a actividade da doença e as necessidades do doente.
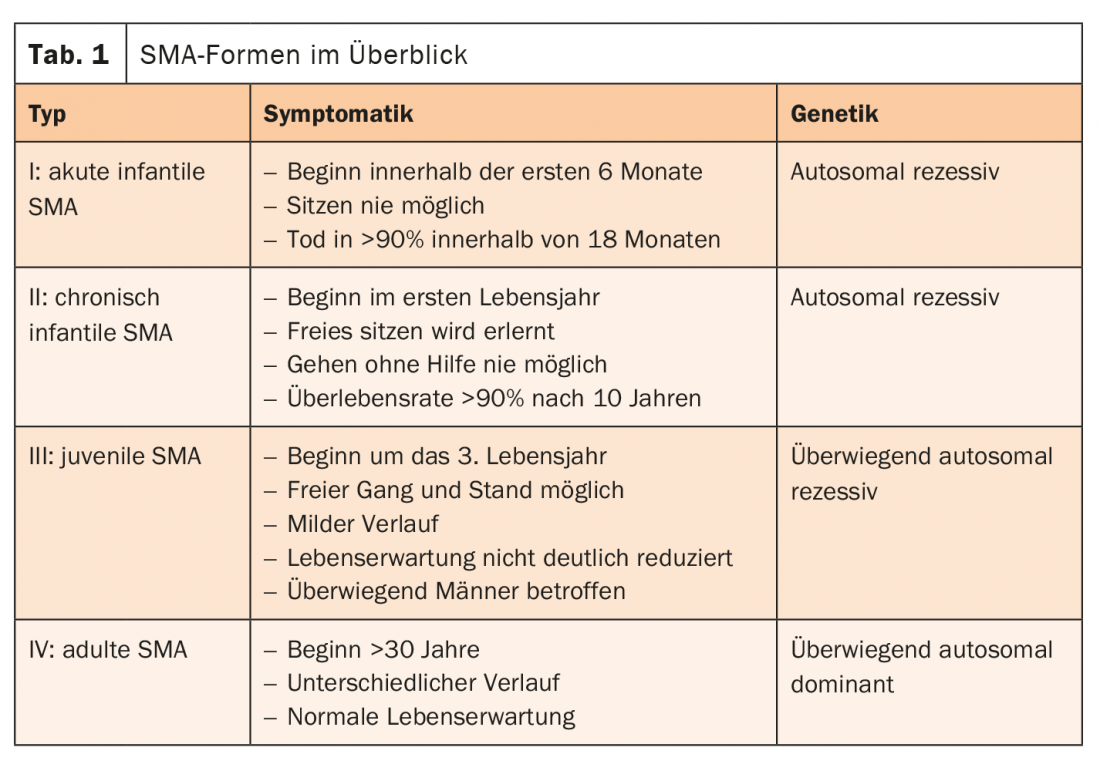
A causa da SMA é geralmente um defeito no gene SMN1. Juntamente com o gene SMN2, forma a proteína “Survival of Motor Neuron” (SMN). Isto desempenha um papel central na transmissão de impulsos das células nervosas para os músculos. Se o gene SMN1 falhar, a proteína importante só pode ser produzida pelo restante gene SMN2. Portanto, quanto mais cópias de SMN2 houver, mais tarde o início do SMA e mais favoravelmente o curso. Os doentes com AME tardia (tipo II-IV) têm frequentemente uma esperança de vida normal. As formas de SMA são diferenciadas de acordo com o padrão de distribuição, início da doença, gravidade e padrão de herança (Tab. 1) [4].
Ecrã de recém-nascidos, se possível
Um diagnóstico definitivo relativo à AME só pode ser feito através de testes genéticos. No entanto, como a AME de tipo 1, se não for tratada, leva à morte ou requer ventilação mecânica permanente em 90% dos casos quando o paciente atinge a idade de dois anos, é indicada a detecção precoce da doença com o início do tratamento mais rápido possível. A gestão terapêutica é complexa e inclui cuidados agudos, bem como medidas relativas à reabilitação, ortopedia, apoio respiratório, fisioterapia e intervenções medicamentosas.
Sucessos terapêuticos também com adolescentes e adultos
O primeiro medicamento que não só actua de forma sintomática mas também aborda as causas da doença através da correcção do defeito genético subjacente foi aprovado em 2017 com nusinersen. O oligonucleotídeo antisense (ASO) é um modulador de emendas específico que deixa o genoma tal como está e melhora a função natural da proteína SMN2. Isto permite a formação de maiores quantidades de proteína SMN completa e funcional. Agora, os primeiros dados de um estudo de coorte multicêntrico foram apresentados. O principal objectivo deste estudo observacional não intervencionista é investigar os objectivos e as expectativas de tratamento de pacientes adultos 5q-SMA, bem como a satisfação subjectiva do tratamento dos pacientes [5].
Os resultados preliminares mostram que os objectivos de tratamento individual são de particular importância. Dentro dos diferentes tipos de 5q-SMA, os objectivos da terapia variam significativamente. Assim, a preservação da função do braço é um dos objectivos de tratamento mais comuns, com predominância em doentes de tipo 1 e tipo 2. Nos doentes com AME tipo 3, a preservação e melhoria da função das pernas é mais frequentemente priorizada. A terapia ASO parece ser adequada para isto. Resultados de outros estudos mostram também que várias capacidades motoras podem estabilizar ou mesmo melhorias motoras clinicamente relevantes são possíveis em adultos com 5q-SMA [6-8].
Congresso: DGM 2021
Literatura:
- Bowerman M, et al: Estratégias terapêuticas para a atrofia muscular espinhal: SMN e mais além. Dis Modelo Mech 2017; 10: 943-954.
- Borasio G, et al: Diagnóstico de atrofias musculares espinais. Neurologia 2001; 20:113-118.
- www.sma-schweiz.ch/spinale-muskelatrophie/typen-der-proximalen-sma (último acesso 05.04.2021)
- www.muskelgesellschaft.ch/diagnosen/spinale-muskelatrophien-sma (último acesso 05.04.2021)
- Meyer, et al: Congresso DGN 2020, P333.
- Hagenacker T, et al: Lancet Neurol 2020; 19(4): 317-325.
- Walter MC, et al: J Neuromuscul Dis 2019; 6: 453-465.
- Maggi L, et al: JNNP. 2020, Nov;91(11): 1166-1174.
InFo NEUROLOGY & PSYCHIATRY 2021; 19(3): 41 (publicado 5.6.21, antes da impressão).
PRÁTICA DO GP 2021; 16(8): 45