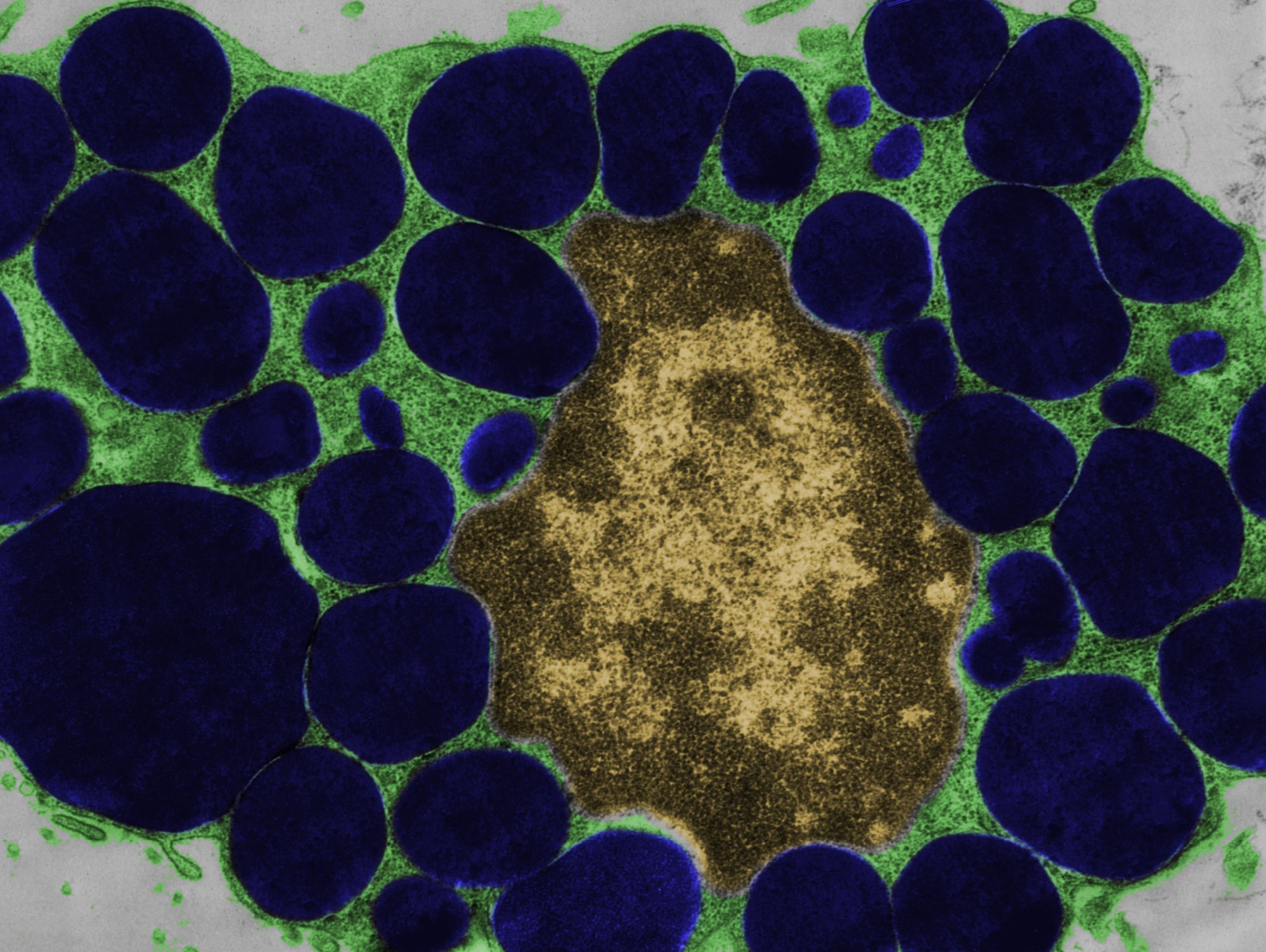
A destruição crónica dos eritrócitos é característica da hemoglobinúria paroxística nocturna. As mutações no gene PIG-A levam a um défice GPI-AP, o que resulta numa activação descontrolada do complemento. A inibição do complemento C5, que só precisa de ser administrada de oito em oito semanas pela primeira vez, pode ajudar.
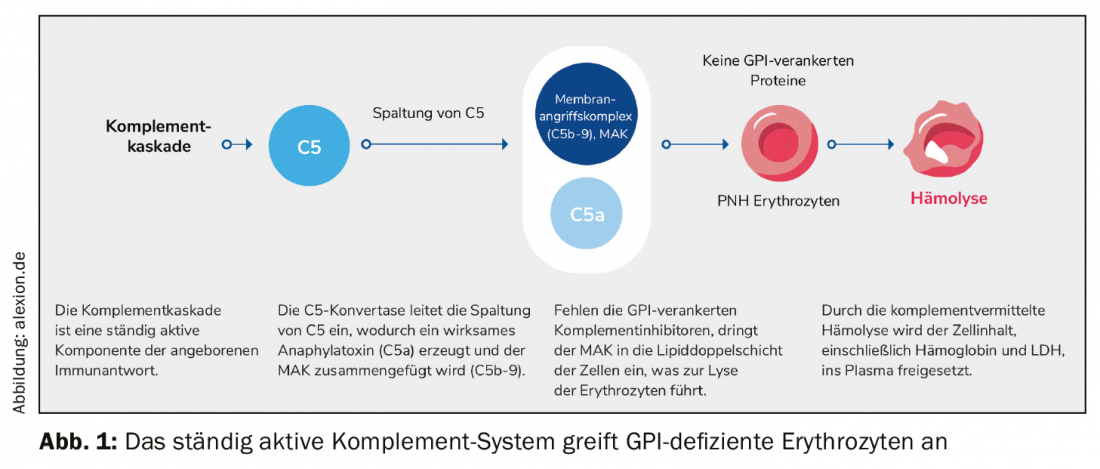
A principal característica da hemoglobinúria nocturna paroxística rara, crónica e progressiva (PNH) é a hemólise completa-mediada. Devido a uma mutação adquirida, faltam proteínas âncora de glicofosfatidilinostiol (GPI) nas células estaminais hematopoiéticas da medula óssea. Como resultado, não é possível a formação de proteínas com eritema GPI, especialmente na superfície de eritrócitos vermelhos. A protecção contra o sistema complemento, uma parte do próprio sistema imunitário do corpo, cai. Os glóbulos vermelhos são confundidos com invasores, atacados e destruídos. O complexo de ataque de membrana (MAK) penetra no bocal lipídico da célula e desencadeia a lise dos eritrócitos que aí se encontram. Todo o conteúdo celular, incluindo hemoglobina e LDH, é libertado para o plasma (Fig. 1).
Doença complexa com sintomas variáveis
A doença caracteriza-se por anemia hemolítica, hemoglobinúria e insuficiência ou falência da medula óssea, entre outros sintomas. Manifesta-se classicamente principalmente pela fadiga, uma qualidade de vida reduzida e dispneia ao esforço. No entanto, também podem ocorrer sintomas não específicos que acompanham frequentemente crises hemolíticas. No entanto, a principal causa do aumento da morbilidade e mortalidade dos pacientes é a trombofilia. Até 50% de todas as pessoas afectadas desenvolvem tromboses sem medidas terapêuticas específicas. 35% dos doentes morrem no prazo de 5 anos após o diagnóstico.
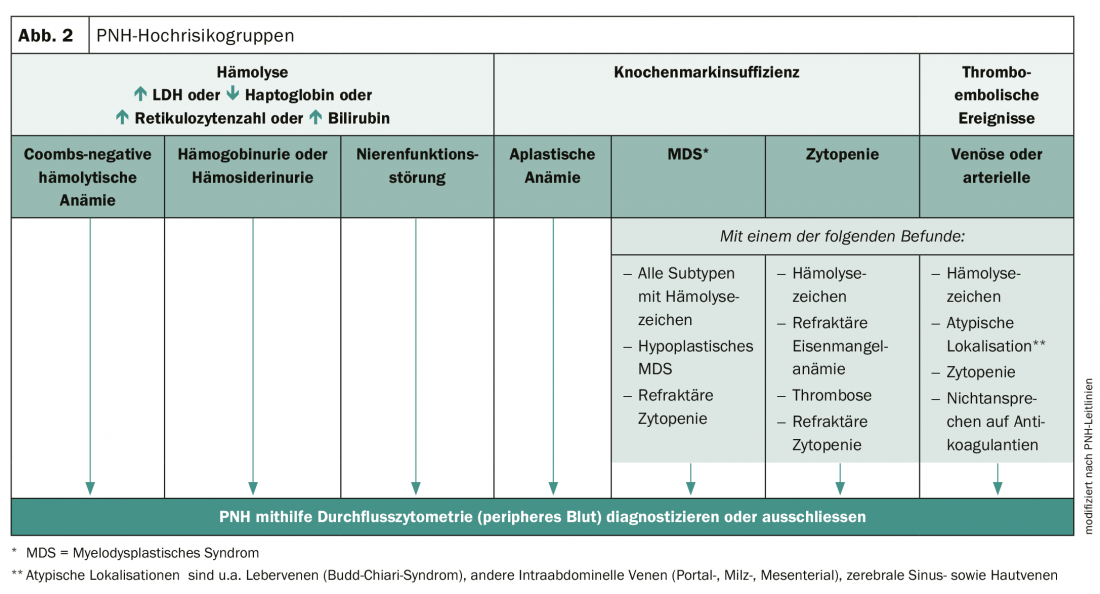
O diagnóstico precoce é essencial para melhorar o prognóstico das pessoas afectadas. A PNH pode ser diagnosticada usando citometria de fluxo de alta sensibilidade e um exame clínico abrangente. No entanto, muitos médicos não estão conscientes da complexidade dos sintomas e da variabilidade da doença. Como resultado, a PNH não é frequentemente reconhecida e o diagnóstico só é feito após um atraso de um a mais de 5 anos. Contudo, existem grupos de alto risco em que a presença da PNH deve ser considerada (Fig. 2).
A gestão terapêutica melhora
Para parar a hemólise, as funções terminais da cascata do complemento são interferidas por meio de inibidores do complemento. As funções proximais, por outro lado, permanecem. Por exemplo, a taxa de eventos tromboembólicos (TE) foi significativamente reduzida de 11,54 para 0,72 em doentes tratados com anticoagulantes. O inibidor foi também convincente em termos de benefício clínico a longo prazo. O único inconveniente: o curto intervalo de infusão. Agora o inibidor poderia ser mais desenvolvido e a meia-vida prolongada (Ravulizumab). Ao reduzir o número de infusões para 6-7 por ano, a qualidade de vida das pessoas afectadas é significativamente melhorada e o risco de hemólise revolucionária também foi reduzido de 10,7% para 4%.
Fonte: Reunião Anual 2019 das Sociedades de Hematologia e Oncologia Médica de Língua Alemã (DGHO)
InFo ONCOLOGY & HEMATOLOGY 2019; 7(6): 32-33 (publicado 6.12.19, antes da impressão).