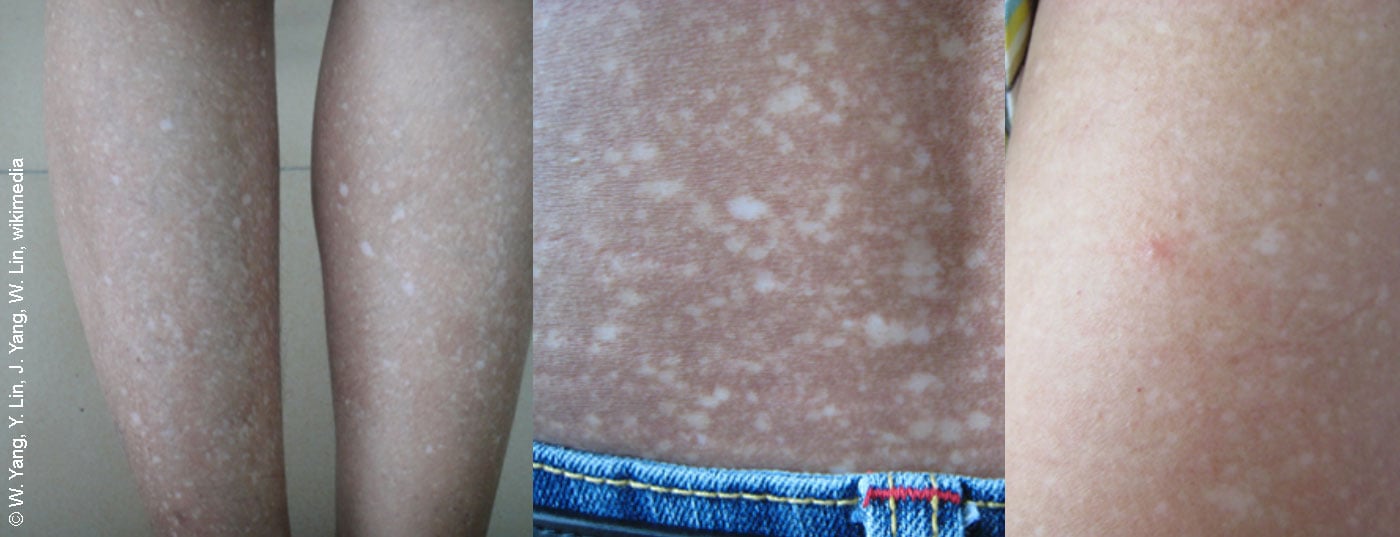
A amiloidose é uma forma rara de dermatose de depósito causada pela dobra incorrecta das proteínas. A detecção precoce dermatológica é de grande importância para a continuação do curso da doença.
Etimologicamente, o termo amiloidoses remonta ao grego antigo ἄμυλον ámylon, que significa “refeição de poder, amido”. Uma vez que bioquimicamente está presente uma estrutura proteica, este é um nome enganador [1]. Os amilóides são um grupo de proteínas heterogéneas que aparecem de forma ultra-estrutural semelhante ou reagem de forma semelhante na coloração histológica. Tanto as amiloidoses localizadas como sistémicas podem ser caracterizadas por sintomas cutâneos. “Mais de 25 proteínas são capazes de formar amilóide em pessoas saudáveis”, disse o Dr. med. Antonio Cozzio, Clínica de Dermatologia, Venereologia e Alergologia do Hospital Cantonal St.Gallen, que falou sobre este tema nos Dias de Formação Dermatológica de Zurique deste ano. Das proteínas bioquimicamente diferentes que são capazes de formar amilóide, 15 podem levar a doenças clinicamente relevantes [2].

Mudança de taxonomia
A detecção precoce pelo dermatologista é uma base importante para o tratamento eficaz e a prevenção da deterioração posterior de quaisquer órgãos envolvidos [3]. Em 2016, o sistema de classificação da Sociedade para Amiloidoses foi revisto [4]: A congofilia e a birefringência continuam a ser os critérios classificatórios mais importantes. Além disso, é recomendado determinar a identidade química da proteína fibrilosa amilóide depositada no espaço extracelular dos tecidos através da análise da sequência proteica. Até à data, são conhecidos no homem 36 teins fibrilares extracelulares, 2 dos quais são iatrogénicos por natureza, 9 também foram identificados em animais. Duas proteínas fibrilares recentemente descobertas são AApoCII (derivado da apolipoproteína CII) e AApoCIII (derivado da apolipoproteína CIII). AApoCII e AApoCIII amiloidose são amiloidoses sistémicas hereditárias. Duas proteínas anteriormente consideradas inclusões intracelulares, tau e α-synuclein, são agora classificadas como depósitos extracelulares associados à morte celular, designadas “ATau” e “AαSyn”.
Envolvimento cutâneo heterogéneo
É feita uma distinção entre formas de amiloidose sistémica e cutânea limitada, cada uma das quais subdividida em subtipos adquiridos e hereditários (Fig. 1).
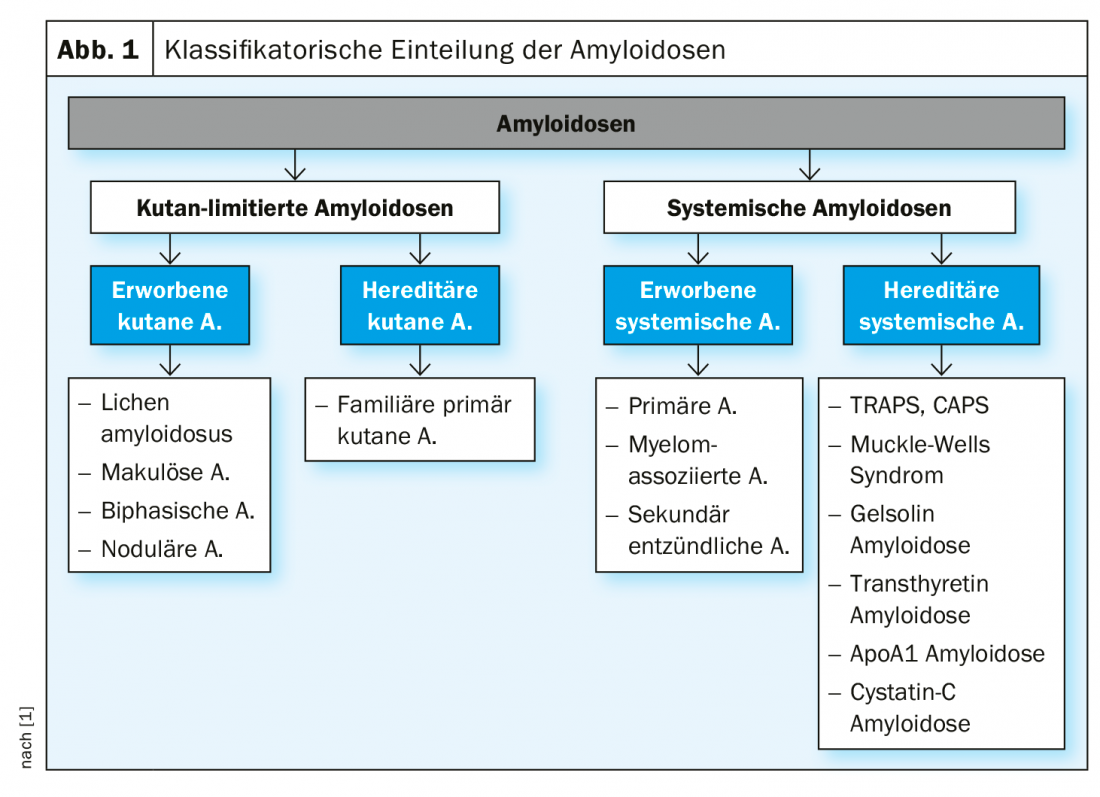
Amiloidoses sistémicas: Este é um quadro clínico heterogéneo que se caracteriza pela deposição das proteínas do próprio corpo alteradas patologicamente nos tecidos ou órgãos. Os sinais cutâneos característicos das diferentes formas de amiloidose sistémica adquirida estão resumidos no Quadro 1.
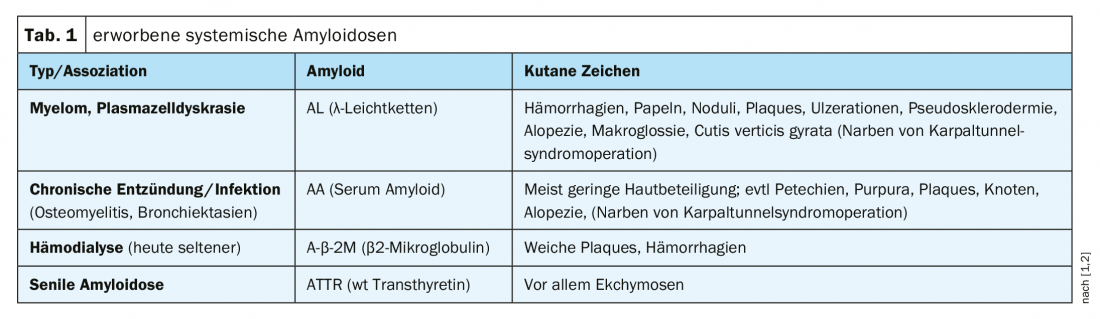
A amiloidose da cadeia ligeira é a forma mais comum e é frequentemente muito agressiva. Se o coração ou os rins forem afectados, esta forma de amiloidose pode levar à morte após alguns meses se não for tratada. Pensa-se que as cadeias leves de anticorpos desempenham um papel importante na etiologia e conduzem aos depósitos característicos no tecido. A amiloidose sistémica é comum em doentes com mieloma múltiplo. Numa fase inicial de amiloidose sistémica primária, as manifestações clínicas limitam-se inicialmente aos sintomas dermatológicos; o envolvimento de órgãos ocorre apenas no curso posterior [5]. O espectro de diagnóstico inclui biopsia de tecidos e medula óssea, electroforese e exames específicos de órgãos (visão geral 1).


Amiloidoses cutâneas limitadas: Este termo colectivo descreve patologias dermatológicas que são histologicamente caracterizadas pela acumulação extracelular de depósitos amilóides na derme. “As amiloidoses macular e papular são normalmente limitadas”, explica o orador. Se houver suspeita de amiloidose cutânea nodal, deve ser esclarecido se existe envolvimento cardíaco/renal; pode ser necessária a cooperação com a hemato-oncologia [1]. No caso de púrpura pouco clara (especialmente no caso de formação de nódulos adicionais, macroglossia), recomenda-se uma biópsia com análise imuno-histoquímica, onde também podem ser incluídos os achados histológicos da história anterior. Devem ser excluídos os seguintes diagnósticos diferenciais: neoplasia cutânea, sarcoidose, granuloma facial, leishmaniose (visão geral 2) [1]. O espectro de opções de tratamento é relativamente amplo e deve ser adaptado individualmente (caixa).
Literatura:
- Cozzio A: Apresentação de slides: Tema anual das dermatoses deposicionais. Amiloidoses, Antonio Cozzio, MD, 9th Zurich Dermatology Training Days 2019, Zurique, 26 de Junho de 2019.
- Rauch PJ: Amiloidoses sistémicas. Swiss Med Forum 2014; 14(50): 943-948.
- Bruch-Gerharz D, Ruzicka T: Amiloidoses e hialinoses. In: Plewig G., Ruzicka T., Kaufmann R, Hertl M (eds) Braun-Falco’s Dermatology, Venereology and Allergology 2017. Medicina de Referência Springer. Berlim, Heidelberg: Springer.
- Sipe JD, et al: Amyloid fibril proteins and amyloidosis: chemical identification and clinical classification International Society of Amyloidosis 2016 Nomenclature Guidelines. Amilóide. The Journal of Protein Folding Disorders
- Trajber Horvat A, Trčko K, Jurčić V, Marko PB: Primary Systemic Amyloidosis With Skin and Cardiac Involvement: A Case Report. Acta Dermatovenerologica Alpina, Pannonica, et Adriatica 2018. www.oneamyloidosisvoice.com/rcuratenew
DERMATOLOGIE PRAXIS 2019; 29(5): 27-28 (publicado 10.10.19, antes da impressão).