Para além dos cuidados habituais para todas as formas de ELA, registaram-se progressos significativos, em particular para as formas monogenéticas de ELA, graças ao desenvolvimento de métodos terapêuticos específicos baseados em genes, atualmente a ASO em particular. Os diagnósticos genéticos de base, pelo menos dos genes mais comuns (SOD1, C9orf72, FUS, TARDP), são por isso recomendados para todos os doentes com ELA no momento do diagnóstico.
Pode fazer o teste CME na nossa plataforma de aprendizagem depois de rever os materiais recomendados. Clique no botão seguinte:
Os casos de esclerose lateral amiotrófica (ELA) foram descritos pela primeira vez por Jean Martin Charcot em 1873 [1]. Charcot já tinha fornecido provas neuropatológicas de uma degeneração subjacente do sistema motor. Sabe-se agora que a esclerose lateral amiotrófica é uma neurodegenerescência multissistémica com numerosas manifestações extramotoras, apesar da degenerescência predominante do primeiro e segundo neurónios motores e do trato corticoespinal. O conhecimento dos factores genéticos subjacentes tem vindo a aumentar, sobretudo na última década, o que levou também às primeiras consequências terapêuticas diretas.
Epidemiologia
Com base nos dados do registo de doentes mais bem gerido na Suábia, pode estimar-se que existem cerca de 8.000 a 9.000 pessoas afectadas pela ELA em toda a Alemanha [2]. Com uma idade média de início de 70 a 75 anos e uma ligeira predominância masculina, assume-se uma incidência de aproximadamente 3/100.000 doentes. A prevalência ao longo da vida como a medida estatística mais descritiva para a probabilidade de desenvolver ELA é de 1:400. Estes valores epidemiológicos são altamente congruentes com os valores de outros países europeus. [3,4]Noutras partes do mundo, como a Ásia, os dados epidemiológicos são diferentes.
Sintomas clínicos
Os principais sintomas da ELA são paresia atrófica, progressiva e de início focal, com espasmos e fasciculações musculares frequentes, como sinal de danos crescentes nos segundos neurónios motores ao nível da coluna vertebral ou na área do tronco cerebral [5]. Esta situação é precedida ou acompanhada por uma afeção dos primeiros neurónios motores no córtex motor primário e no trato cortico-espinal, com reflexos de estiramento muscular aumentados e saltitantes e evidência de reflexos patológicos ou um aumento do tónus muscular no sentido da espasticidade.
[6,7]Clinicamente e na vida quotidiana, défices cognitivos relevantes e anomalias comportamentais no sentido de uma síndrome de demência frontotemporal são encontrados como sintomas extra-motores em aproximadamente 5% das pessoas afectadas pela ELA. As perturbações do sistema nervoso autónomo que as acompanham têm sido cada vez mais descritas nos últimos anos [8]. Para além disso, a dor de várias origens também é importante durante o curso da doença [9].Procedimentos de diagnóstico e diagnóstico
O diagnóstico é feito principalmente de forma clínica após a exclusão consistente e cuidadosa de diagnósticos diferenciais alternativos. [10]Os critérios da Gold Coast fornecem uma boa orientação para o diagnóstico clínico. O procedimento de diagnóstico de apoio mais importante é a eletromiografia para detetar desnervação ativa ou potenciais de fasciculação politópicos com evidência simultânea de lesão neurogénica crónica. [11]Na eletromiografia, devem ser examinados diferentes músculos das quatro regiões do corpo (nervos cranianos/bulbar – cervical – torácico – lombossacral). Além da eletromiografia, a ecografia muscular pode também ser realizada para determinar a presença de fasciculações musculares politópicas e para avaliar o trofismo muscular e a estrutura interna. A ultrassonografia muscular pode ser particularmente vantajosa para a deteção de fasciculações musculares em músculos maiores ou mais profundos, como partes do músculo quadríceps femoral ou músculos da base da língua, e pode ser um complemento importante da eletromiografia. A electroneurografia dos nervos motores e sensoriais, bem como o diagnóstico da onda F nos membros superiores e inferiores, são particularmente necessários para excluir a neuropatia desmielinizante primária e, em particular, os bloqueios de condução (PDIC ou neuropatia motora multifocal/NMF).
A estimulação magnética transcraniana para o registo dos potenciais evocados motores está disponível para objetivar e, se necessário, quantificar o envolvimento dos primeiros neurónios motores. [12]Este método de exame pode ser utilizado para examinar a condução motora central dos axónios espessamente mielinizados do trato corticoespinal a partir do córtex motor como local de estimulação ao longo de toda a medula espinal. Faz sentido examinar um músculo distal em cada extremidade superior e inferior. O quadro 1 apresenta uma panorâmica pormenorizada dos diagnósticos electrofisiológicos propostos para a ELA e dos critérios de diagnóstico da Gold Coast .
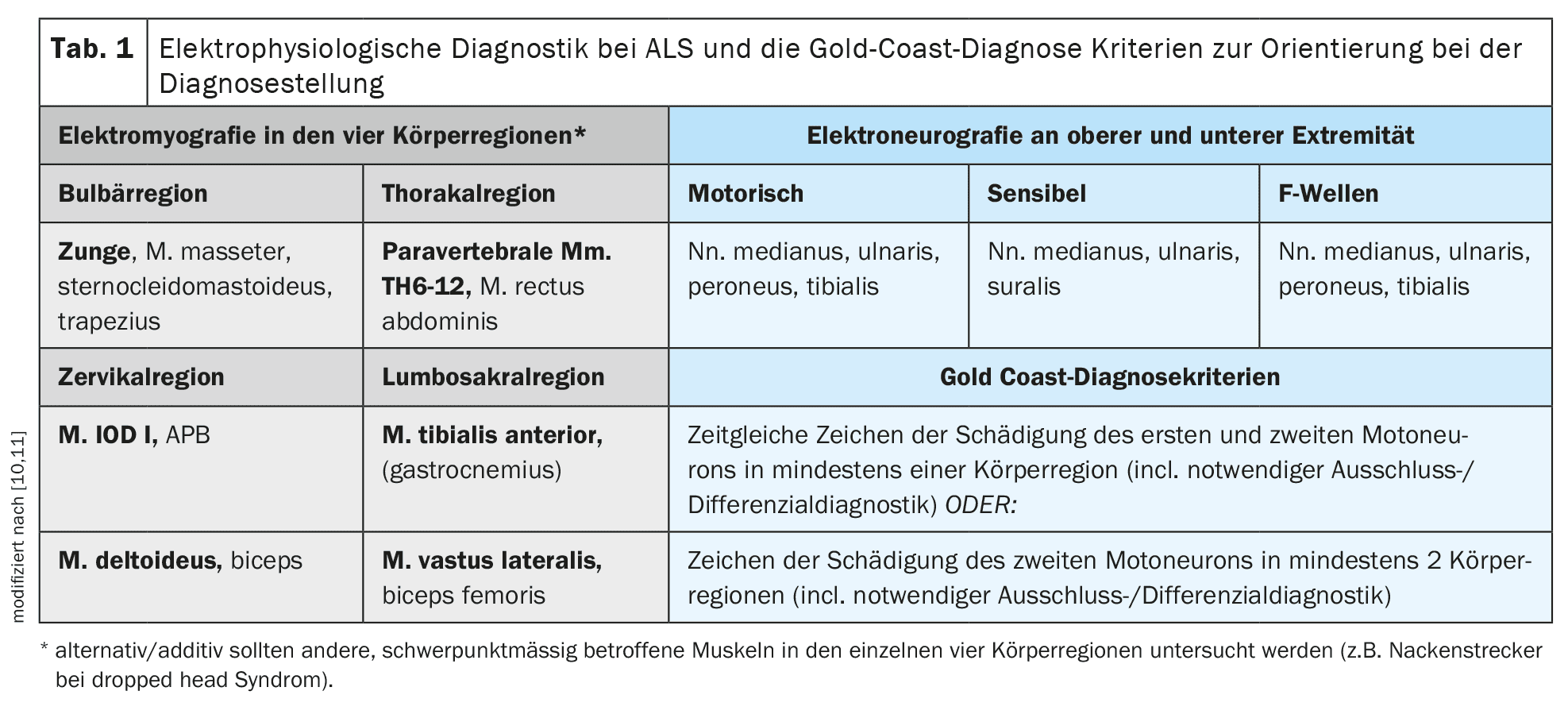
Embora os diagnósticos genéticos ainda fossem considerados opcionais na diretriz S1, que é válida e está disponível desde 2021, os autores acreditam que esta situação mudou em resultado dos desenvolvimentos dos últimos três anos [5]. [15,16]Tendo em conta os desenvolvimentos terapêuticos neste domínio, todos os doentes com ELA devem ser submetidos a testes genéticos orientados obrigatórios, pelo menos para a presença de uma mutação no gene Cu/Zn superóxido dismutase 1(SOD 1) e os doentes jovens com ELA com menos de 40 anos para a presença de uma mutação patogénica no gene FUS. [17]Opcionalmente, devem também ser efectuados diagnósticos genéticos mais extensos com o exame de outros genes, como o C9orf72, TARDP, TBK1, etc. . As secções seguintes, relativas à etiologia e à genética e, em particular, à terapêutica, abordarão estas questões com mais pormenor. O Quadro 1 apresenta uma visão geral dos procedimentos de diagnóstico descritos e dos critérios de diagnóstico da Gold Coast recomendados como guia na prática clínica .
No que diz respeito a outras investigações clinicamente relevantes para diagnósticos diferenciais e de exclusão abrangentes, dependendo da apresentação clínica inicial (por exemplo, imagiologia por RM, diagnósticos ORL, diagnósticos laboratoriais), bem como diagnósticos relevantes durante o curso da doença para avaliar o prognóstico (por exemplo, teste de função pulmonar/teste de função diafragmática, diagnóstico da deglutição utilizando o FEES, psicometria utilizando o ECAS), gostaríamos de remeter explicitamente para a apresentação muito pormenorizada e clara da diretriz S1 [5].
Espectro fenotípico e padrão de paresia muscular no contexto da neuroanatomia e fisiopatologia da ELA
As descobertas científicas das últimas duas décadas, em particular os estudos neuroanatómicos e neuropatológicos, alteraram fundamentalmente a visão da ELA. [18,19]Atualmente, a ELA já não é vista como uma degenerescência puramente do sistema motor, mas como uma degenerescência multissistémica.
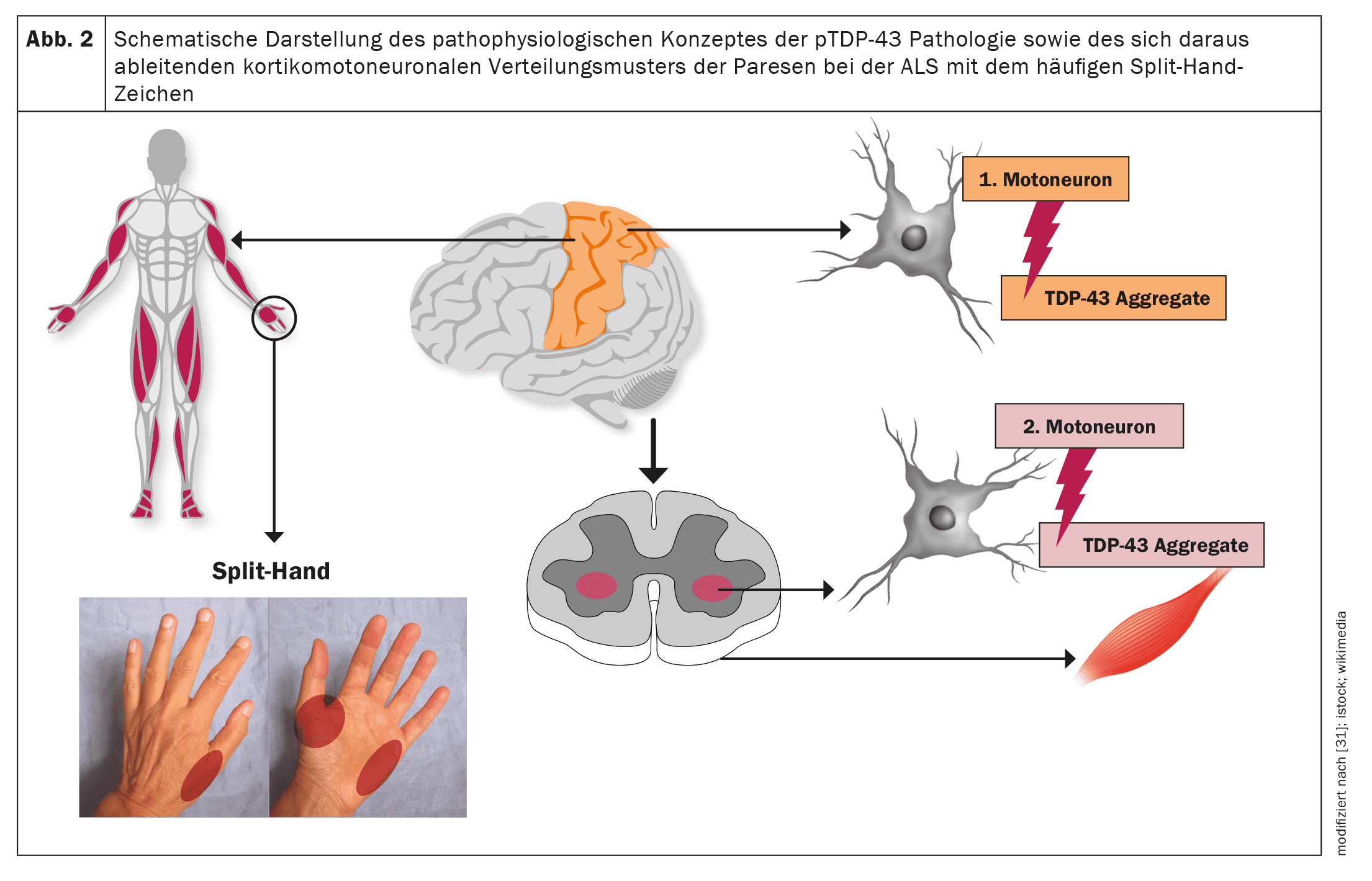
A ELA, tal como outras doenças neurodegenerativas, como a doença de Alzheimer ou a doença de Parkinson, é uma proteinopatia, ou seja, os agregados proteicos patológicos nas células nervosas motoras, como correlato neuropatológico, conduzem à disfunção das células nervosas motoras afectadas e, em última análise, à apoptose e, consequentemente, à perda de células nervosas motoras. [20]No caso da ELA, mais de 95% dos casos envolvem agregados da proteína TDP-43. [21]Só no caso de variantespatológicas subjacentes da SOD1 ou da FUSé que ocorre uma neurodegenerescência independente da TDP-43. [22]Estudos neuropatológicos estabeleceram uma propagação cerebral gradual e faseada da patologia TDP-43 na ELA, comparável à patologia da α-sinucleína na doença de Parkinson e à patologia da tau na doença de Alzheimer. Estes achados, bem como o padrão corticomotoneuronal de paresia com o sinal da mão dividida (atrofia assimétrica dos músculos C8/T1 ou [23–26](atrofia assimétrica dos músculos C8/T1 ou ulnar da mão) como um sinal clínico muito comum na ELA, implicam uma origem das alterações neuropatológicas na área do córtex motor primário e uma disseminação gradual da patologia TDP-43 a partir daí através do transporte axonal para os segundos neurónios motores, ou seja, os núcleos motores dos nervos cranianos no tronco cerebral e as células do corno anterior da medula espinal.
[27,28]Parece ocorrer uma propagação das alterações neuropatológicas do tipo prião, o que explicaria o início focal da manifestação motora e uma disseminação gradual para miótomos e regiões corporais vizinhas. [29]Assim, dependendo da manifestação inicial da neuropatologia, o espetro fenotípico da ELA – no que diz respeito apenas aos sintomas motores – não é uniforme, mas altamente variável. Por exemplo, podem ocorrer fenótipos motores topograficamente distintos. Além disso, o fenótipo é influenciado pela velocidade geralmente diferente de degeneração do primeiro e segundo neurónios motores e pelos sintomas clínicos associados. Sem uma compreensão precisa dos mecanismos moleculares até à data, parece ser decisiva a relação entre os agregados de proteína TDP-43 insolúveis, que já não podem ser transportados axonalmente e, por conseguinte, se acumulam localmente, e os oligómeros TDP-43 solúveis, precursores dos agregados TDP-43. [23]Obviamente, estes precursores solúveis são depois transportados por via axonal dos primeiros neurónios motores para os segundos neurónios motores, onde são obrigatoriamente depositados como agregados proteicos e conduzem à morte destas células.Até à data, não existe uma classificação normalizada dos fenótipos clínicos que tenha em conta a proporção ou o foco de lesão do primeiro e do segundo neurónio motor, o padrão clínico ou o foco da paresia atrófica e a sua propagação.
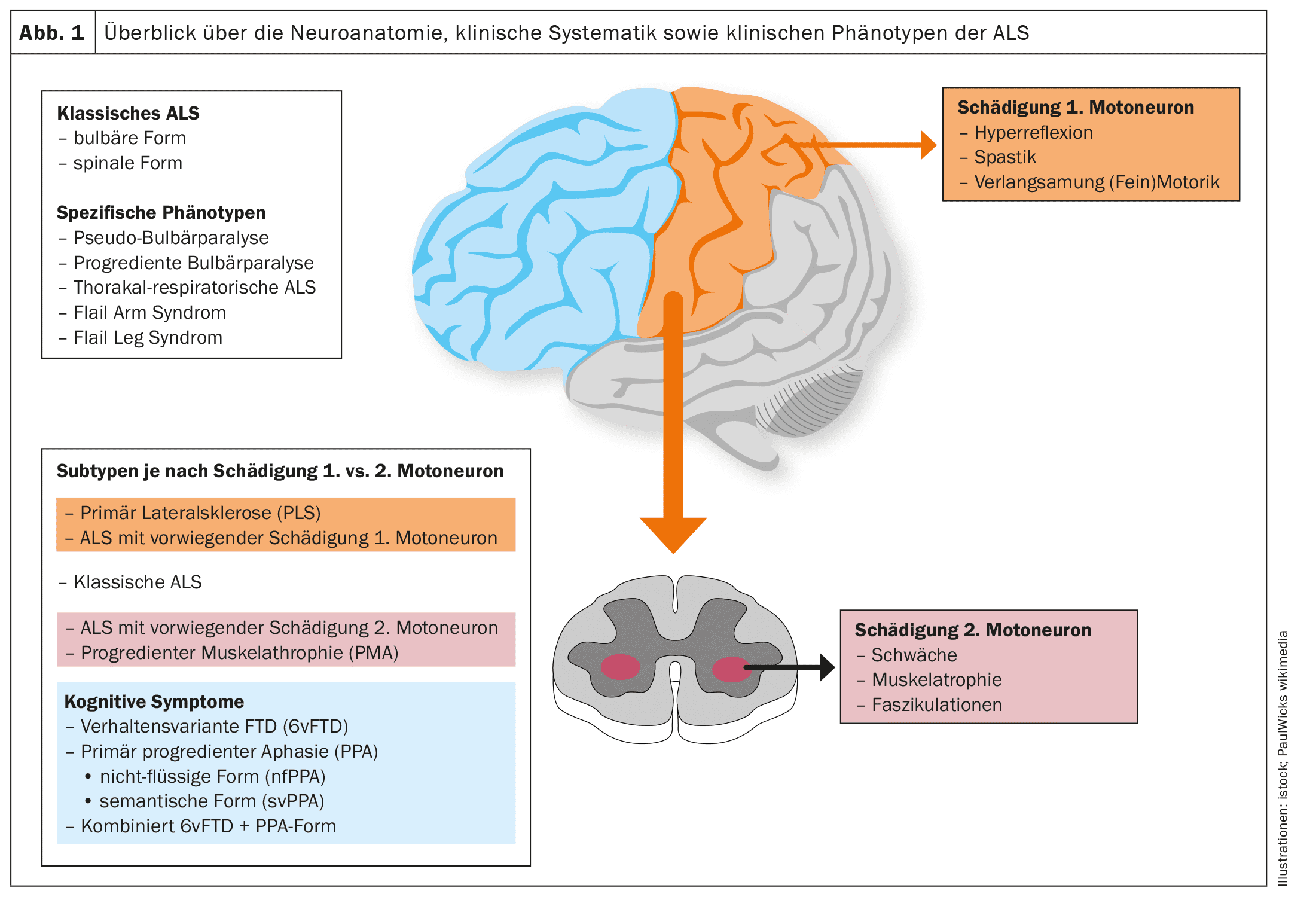
As figuras 1 e 2 fornecem uma visão esquemática da neuroanatomia e da fisiopatologia da doença, bem como dos fenótipos clínicos e da sua classificação.
Etiologia e genética
A etiologia da ELA ainda não é suficientemente bem compreendida. Na forma esporádica da doença, que representa a grande maioria dos casos, assume-se uma génese multifatorial com a interação de vários factores ambientais externos desfavoráveis, factores metabólicos epigenéticos e uma possível suscetibilidade genética adicional. [31]As alterações no transporte proteico nuclear-citosólico, no metabolismo do ARN, na função celular oxidativa mitocondrial, na excitabilidade glutamatérgica, no transporte proteico axonal e na autofagia celular são discutidas como os mecanismos metabólicos intracelulares subjacentes, enquanto as perturbações funcionais dos astrócitos e dos oligodendrócitos, bem como os processos inflamatórios, são discutidos como mecanismos extracelulares aditivos. Foram obtidos conhecimentos significativos sobre os possíveis mecanismos etiológicos da ELA esporádica, principalmente através de formas genéticas com processos fisiopatológicos distintos.
[32]Em 1993, foram descritas pela primeira vez mutações patogénicas na Cu/Zn superóxido dismutase 1 (SOD1) como causa genética da ELA. Atualmente, sabe-se que um grande número de mutações genéticas são a etiologia subjacente da ELA. [17]No caso de ELA aparentemente esporádica sem história familiar evidente na Alemanha, pode ser detectada uma causa monogenética em pouco mais de 10% dos casos. [17]As causas genéticas mais comuns na Alemanha são as expansões patológicas da repetição C9orf72 e as mutações nos genes SOD1, TARDP e FUS.O diagnóstico genético no momento do diagnóstico deve, portanto, ser oferecido a todos os doentes com ELA, uma vez que pode ter consequências terapêuticas imediatas, que discutiremos em pormenor na secção seguinte.
Terapia e prognóstico
O Riluzole foi aprovado na Alemanha desde 1996 como a única substância farmacológica com um efeito positivo comprovado na progressão da ELA. Postula-se como mecanismo de ação decisivo uma redução da libertação de glutamato e, consequentemente, uma redução da excitotoxicidade. [18]Esta hipótese foi reforçada pela evidência de uma degenerescência primária das vias glutamatérgicas corticofugais. Uma dose diária de 100 mg de riluzol apresenta o melhor perfil de eficácia/efeitos secundários. [33–35]As análises retrospectivas de um total de dez registos clínicos de ELA fornecem provas de um prolongamento médio do tempo de sobrevivência até 19 meses com o riluzol, havendo também indicações claras de que esta substância também é eficaz em fases mais avançadas da doença. O riluzol é geralmente bem tolerado; os efeitos secundários conhecidos incluem um aumento das transaminases, que devem, por isso, ser monitorizadas regularmente, e queixas gastrointestinais. Atualmente, estão também disponíveis formas de dosagem alternativas, como sumos ou comprimidos orodispersíveis, para doentes com ELA com disfagia.
Para além da terapêutica farmacológica com riluzol, considera-se que a prevenção de um estado metabólico catabólico com uma perda de peso consecutiva tem um benefício prognóstico adicional. [36]Quanto mais elevado for o índice de massa corporal (IMC), mais favorável é o prognóstico.
As recomendações actuais para os doentes com ELA são, portanto, manter um peso estável e evitar a perda de peso. [37]Neste contexto, a inserção de um tubo PEG também desempenha um papel importante na disfagia progressiva, com um tempo de sobrevivência prolongado em resultado desta medida. A questão de saber se, e em que medida, abordagens terapêuticas anti-catabólicas específicas, tais como intervenções nutricionais específicas com elevado teor de gordura e de calorias ou intervenções nutricionais cetogénicas, podem melhorar o prognóstico está atualmente a ser intensamente investigada em estudos. Neste contexto, o estudo LIPCAL-ALS 2 a nível alemão como Investigator Initiated Trial (IIT) por colegas de Ulm, com início previsto para 2024 a 2025, é de grande importância.
[38,39]A ventilação não invasiva (VNI) e invasiva por meio de um traqueostoma é outra medida importante para prolongar o tempo de sobrevivência na ELA com insuficiência ventilatória. Isto também é plausível, uma vez que a hipoventilação alveolar com hipercapnia consecutiva devido ao envolvimento diafragmático é tipicamente um fator significativo na morte de doentes com ELA após três a cinco anos de progressão da doença.O desenvolvimento de oligonucleótidos antisense (ASO) para formas específicas de ELA induzidas geneticamente pode ser visto como um marco. Em particular, deve ser mencionado aqui o ASO Tofersen, administrado por via intratecal, que se liga ao ARNm da SOD1em doentes com variantespatogénicas da SOD1(cerca de 1-2% de todos os casos de ELA), impedindo assim a expressão citotóxica da proteína SOD1. [16,40]Foi demonstrado que o efeito mecanicista de uma redução significativa da expressão da SOD1em cerca de 30% nos seres humanos ocorre muito rapidamente nos dias seguintes ao início da terapêutica com Tofersen, seguido de uma queda significativa do NfL no LCR e no soro, antes de ocorrer finalmente um abrandamento do declínio da pontuação ALSFRS-R. Após uma observação a longo prazo de doze meses, foram também observados outros sinais positivos com relevância clínica, tais como efeitos na função muscular e estabilização do peso.
Nos EUA, o Tofersen foi aprovado pela FDA em abril de 2023 apenas com base nos dados convincentes dos biomarcadores com uma queda significativa nos valores NfL. Na Alemanha, o Tofersen foi disponibilizado como parte de um programa de acesso aberto. [41]Os primeiros dados de aplicação no mundo real da rede alemã de neurónios motores confirmaram de forma impressionante os dados do estudo do Tofersen com dados ainda melhores em termos de parâmetros de progressão clínica. Neste contexto, foi apenas lógico que a EMA tenha decidido a favor da aprovação do Tofersen em fevereiro de 2024.
[15,42]Para além do ASO Tofersen para a ELA associada à SOD1, estão atualmente a ser desenvolvidos ou já foram investigados em estudos ASOs, sobretudo contra o C9orf72 e o FUS . Em particular, o ASO Jacifusen (ION363) deve ser mencionado aqui quando umavariante FUS patogénica é detectada como a causa da ELA juvenil, que está atualmente a ser testada num estudo multicêntrico e multinacional com dois locais de estudo (Rostock e Ulm) na Alemanha. [43]A ALS-Functional Rating Scale (ALSFRS-R) revista é uma pontuação comprovada e bem estabelecida para avaliar as funções motoras das quatro regiões do corpo e não é apenas um parâmetro importante para estudos clínicos, mas também provou ser um parâmetro de acompanhamento facilmente exequível na prática clínica. [44]Espera-se que a ALSFRS-SE (SE: “self-explanatory”), que só recentemente foi acordada na rede alemã de MND em alemão, com as correspondentes explicações concretas e exemplares para os itens individuais e para as deficiências funcionais, tenha uma vantagem adicional em termos de manuseamento prático e, em particular, de precisão de diagnóstico.Conclusão para a prática
Para além dos cuidados habituais para todas as formas de ELA (Riluzole, prevenção do estado metabólico catabólico, incluindo a colocação atempada de um tubo PEG, ventilação precoce não invasiva e, se necessário, invasiva, em caso de insuficiência ventilatória), registaram-se progressos significativos, sobretudo para as formas monogenéticas de ELA, graças ao desenvolvimento de métodos terapêuticos específicos baseados em genes, atualmente, em particular, o ASO. De referir aqui o Tofersen como uma terapia específica muito eficaz para a ELA associada à SOD1, atualmente também disponível na Alemanha.
Os diagnósticos genéticos básicos, pelo menos dos genes mais comuns (SOD1, C9orf72, FUS, TARDP), são por isso recomendados para todos os doentes com ELA no momento do diagnóstico. A medida em que intervenções específicas, anti-catabólicas e de alto teor calórico podem influenciar favoravelmente o curso da ELA esporádica será, esperamos, demonstrada por estudos futuros nesta área.
Mensagens para levar para casa
- O Riluzole 100 mg/d, a prevenção de um estado metabólico catabólico, incluindo a inserção atempada de um tubo PEG e a ventilação não invasiva precoce em caso de insuficiência respiratória, estão disponíveis como cuidados padrão para todas as formas de ELA. A colocação atempada de um tubo PEG, bem como a ventilação precoce não invasiva e, se necessário, invasiva para a insuficiência ventilatória.
- Além disso, registaram-se progressos significativos, nomeadamente no que diz respeito às formas monogenéticas da ELA. A base para tal é o desenvolvimento de métodos terapêuticos específicos baseados em genes, como os oligonucleótidos antisense (ASO). O Tofersen deve ser mencionado aqui como uma terapia muito eficaz e específica para a ELA associada à SOD1.
- O diagnóstico genético básico, pelo menos dos genes mais comuns (SOD1, C9orf72, FUS, TARDP) é, por isso, recomendado para todos os doentes com ELA no momento do diagnóstico.
Literatura:
- Duyckaerts C, Maisonobe T, Hauw JJ, Seilhean D: Charcot identifica e ilustra a esclerose lateral amiotrófica. Free Neuropathol. 2. doi:10.17879/freeneuropathology-2021-3323.
- Uenal H, Rosenbohm A, Kufeldt J, et al: Incidência e Variação Geográfica da Esclerose Lateral Amiotrófica (ELA) no Sul da Alemanha – Completude do Registo ALS da Suábia. PLoS ONE. 2014; 9(4). doi:10.1371/journal.pone.0093932.
- Jun KY, Park J, Oh KW, et al: Epidemiologia da ELA na Coreia utilizando grandes volumes de dados a nível nacional. J Neurol Neurosurg Psychiatry. 2019;90(4): 395-403. doi:10.1136/jnnp-2018-318974.
- Marin B, Boumédiene F, Logroscino G, et al: Variação na incidência mundial de esclerose lateral amiotrófica: uma meta-análise. Int J Epidemiol 2017; 46(1): 57-74. doi:10.1093/ije/dyw061.
- Petri SA-O GJ Ludolph AC. “Doenças do neurónio motor” da Sociedade Alemã de Neurologia (DGN). (2524-2348; eletrónico).
- Finsel J, Uttner I, Vázquez Medrano CR, et al: Cognição no decurso da ELA – uma meta-análise. Amyotroph Lateral Scler Front Degener. 2023; 24(1-2): 2-13. doi:10.1080/21678421.2022.2101379.
- Iazzolino B, Pain D, Peotta L, et al: Validação da classificação revisada de comprometimento cognitivo e comportamental na ELA. J Neurol Neurosurg Psychiatry 2019; 90(7): 734-739. doi:10.1136/jnnp-2018-319696.
- Oprisan AL, Popescu BO: Disautonomia na Esclerose Lateral Amiotrófica. Int J Mol Sci 2023; 24(19). doi:10.3390/ijms241914927.
- Chiò A, Mora G, Lauria G. Dor na esclerose lateral amiotrófica. Lancet Neurol 2017; 16(2): 144-157. doi:10.1016/S1474-4422(16)30358-1
- Shefner JM, Al-Chalabi A, Baker MR, et al: Uma proposta para novos critérios de diagnóstico para ALS. Clin Neurophysiol 2020; 131(8): 1975-1978. doi:10.1016/j.clinph.2020.04.005.
- Koch JC, Petri S, Zeller D: Diagnóstico Eletrofisiológico de Suspeita de Esclerose Lateral Amiotrófica – Recomendações de Consenso da Rede Alemã de Doenças do Neurónio Motor (MND-NET). Clin Neurophysiol 2024. 2024; 55: 82-88.
- Zoccolella S, Mastronardi A, Scarafino A, et al: Potenciais evocados motores na esclerose lateral amiotrófica: potenciais implicações na deteção do envolvimento subclínico de UMN no fenótipo do neurónio motor inferior. J Neurol 2020; 267(12): 3689-3695. doi:10.1007/s00415-020-10073-5.
- Shahim P, Norato G, Sinaii N, et al. Neurofilaments in Sporadic and Familial Amyotrophic Lateral Sclerosis: A Systematic Review and Meta-Analysis. Genes. 2024;15(4). doi:10.3390/genes15040496.
- Behzadi A, Pujol-Calderón F, Tjust AE, et al: Os neurofilamentos podem diferenciar subgrupos de ALS e ALS de mímicos de diagnóstico comuns. Sci Rep 2021; 11. doi:10.1038/s41598-021-01499-6.
- Korobeynikov VA, Lyashchenko AK, Blanco-Redondo B, et al: Silenciamento da expressão de FUS por oligonucleótidos antisense como abordagem terapêutica na esclerose lateral amiotrófica. Nat Med 2022; 28(1): 104-116. doi:10.1038/s41591-021-01615-z.
- Miller TM, Cudkowicz ME, Genge A, et al: Ensaio de Oligonucleótido Antisense Tofersen para SOD1 ALS. N Engl J Med. Publicado online em 22 de setembro de 2022. doi:10.1056/NEJMoa2204705.
- Ruf WP, Boros M, Freischmidt A, et al: Espectro e frequência de variantes genéticas na esclerose lateral amiotrófica esporádica. Brain Commun 2023; 5(3). doi:10.1093/braincomms/fcad152.
- Braak H, Brettschneider J, Ludolph AC, et al: Esclerose lateral amiotrófica – um modelo de propagação axonal corticofugal. Nat Rev Neurol 2013; 9(12): 708-714. doi:10.1038/nrneurol.2013.221.
- Brettschneider J, Del Tredici K, Toledo JB, et al: Estágios da patologia pTDP-43 na esclerose lateral amiotrófica. Ann Neurol 2013; 74(1): 20-38. doi:10.1002/ana.23937.
- Neumann M: Neuropatologia Molecular das Proteinopatias TDP-43. Int J Mol Sci 2009; 10(1): 232-246. doi:10.3390/ijms10010232.
- Saberi S, Stauffer JE, Schulte DJ, Ravits J: “Neuropatologia da esclerose lateral amiotrófica e suas variantes”. Neurol Clin 2015; 33(4): 855-876. doi:10.1016/j.ncl.2015.07.012.
- Braak H, Braak E: Staging of Alzheimer’s disease-related neurofibrillary changes. Neurobiol Aging 1995; 16(3): 271-278-284.
- Braak H, Ludolph AC: Neumann M, Ravits J, Del Tredici K. As alterações patológicas do TDP-43 nas células de Betz diferem das dos α-motoneurónios bulbares e espinais na esclerose lateral amiotrófica esporádica.
Ata Neuropathol (Berl) 2017; 133(1): 79-90. doi:10.1007/s00401-016-1633-2. - Eisen A, Braak H, Del Tredici K, et al: Influências corticais impulsionam a esclerose lateral amiotrófica. J Neurol Neurosurg Psychiatry 2017; 88 (11): 917-924. doi: 10.1136 / jnnp-2017-315573.
- Ludolph AC, Emilian S, Dreyhaupt J, et al: O padrão de paresia na ELA é consistente com a fisiologia das projeções corticomotoneuronais para diferentes grupos musculares. J Neurol Neurosurg Psychiatry 2020; 91 (9): 991-998. doi: 10.1136 / jnnp-2020-323331.
- Menon P, Kiernan MC, Vucic S: A hiperexcitabilidade cortical precede a disfunção do neurónio motor inferior na ELA. Clin Neurophysiol 2015; 126(4): 803-809. doi:10.1016/j.clinph.2014.04.023.
- Prasad A, Bharathi V, Sivalingam V, et al: Mecanismos Moleculares de TDP-43 Misfolding e Patologia na Esclerose Lateral Amiotrófica. Front Mol Neurosci 2019;12. doi:10.3389/fnmol.2019.00025.
- Gosset P, Camu W, Raoul C, Mezghrani A: Prionóides na esclerose lateral amiotrófica. Brain Commun 2022;4(3). doi:10.1093/braincomms/fcac145.
- Hardiman O, Al-Chalabi A, Chio A, et al: Esclerose lateral amiotrófica. Nat Rev Dis Primer 2017; 3:17071. doi:10.1038/nrdp.2017.71.
- Masrori P, Van Damme P: Esclerose lateral amiotrófica: uma revisão clínica. Eur J Neurol 2020; 27(10): 1918-1929. doi:10.1111/ene.14393.
- Eisen A, Vucic S, Mitsumoto H: História da ELA e as teorias concorrentes sobre a patogénese: capítulo do manual da IFCN. Clin Neurophysiol Pract 2023; 9: 1-12. doi:10.1016/j.cnp.2023.11.004.
- Rosen DR, Siddique T, Patterson D, et al: Mutações no gene da superóxido dismutase Cu/Zn estão associadas à esclerose lateral amiotrófica familiar. Nature 1993; 362(6415): 59-62. doi:10.1038/362059a0.
- Bensimon G, Lacomblez L, Meininger V, Group the AS: A Controlled Trial of Riluzole in Amyotrophic Lateral Sclerosis. http://dx.doi.org/10.1056/NEJM199403033300901.
- Hinchcliffe M, Smith A: Riluzole: evidências do mundo real apoiam uma extensão significativa dos tempos de sobrevivência mediana em pacientes com esclerose lateral amiotrófica. Degener Neurol Neuromuscul Dis 2017;7: 61-70. doi:10.2147/DNND.S135748.
- Miller RG, Mitchell JD, Moore DH: Riluzol para a esclerose lateral amiotrófica (ELA)/doença do neurónio motor (DNM).
Cochrane Database Syst Rev 2012; 2012(3).
doi:10.1002/14651858.CD001447.pub3. - Dupuis L, Pradat PF, Ludolph AC, Loeffler JP: Metabolismo energético na esclerose lateral amiotrófica. Lancet Neurol 2011; 10(1): 75-82. doi:10.1016/S1474-4422(10)70224-6.
- Cui F, Sun L, Xiong J, et al: Efeitos terapêuticos da gastrostomia endoscópica percutânea na sobrevivência em pacientes com esclerose lateral amiotrófica: uma meta-análise. PLoS ONE 2018; 13(2). doi:10.1371/journal.pone.0192243.
- Dorst J, Ludolph AC: Ventilação não invasiva na esclerose lateral amiotrófica. Ther Adv Neurol Disord 2019; 12: 1756286419857040. doi:10.1177/1756286419857040.
- Radunovic A, Annane D, Rafiq MK, et al: Ventilação mecânica para esclerose lateral amiotrófica/doença do neurónio motor.
Cochrane Database Syst Rev 2017; 2017(10). doi:10.1002/14651858.CD004427.pub4. - Miller T, Cudkowicz M, Shaw PJ, et al: Ensaio de Fase 1-2 do Oligonucleótido Antisense Tofersen para SOD1 ALS. N Engl J Med 2020; 383(2): 109-119. doi:10.1056/NEJMoa2003715.
- Wiesenfarth M, Dorst J, Brenner D, et al: Efeitos do tratamento com tofersen em doentes com SOD1-ALS num contexto do “mundo real” – um estudo de coorte multicêntrico de 12 meses do programa alemão de acesso antecipado. eClinicalMedicine. 2024; 69. doi:10.1016/j.eclinm.2024.102495.
- Meijboom KE, Brown RH: Abordagens à terapia de modulação genética para a ELA. Neurotherapeutics 2022;19(4): 1159-1179. doi:10.1007/s13311-022-01285-w.
- Cedarbaum JM, Stambler N, Malta E, et al: A ALSFRS-R: uma escala de classificação funcional da ELA revista que incorpora avaliações da função respiratória. Grupo de Estudo BDNF ALS (Fase III). J Neurol Sci 1999; 169(1-2): 13-21. doi:10.1016/s0022-510x(99)00210-5.
- Maier A, Boentert M, Reilich P, et al: ALSFRS-R-SE: uma versão adaptada, anotada e auto-explicativa da escala de classificação funcional da esclerose lateral amiotrófica revista. Neurol Res Pract 2022; 4. doi:10.1186/s42466-022-00224-6.
| Publicado pela primeira vez em neuro aktuell 2024; 38(8): 30-35. |
InFo NEUROLOGY & PSYCHIATRY 2024; 22(4): 6-11