As dermatoses auto-imunes bolhosas representam um grupo heterogéneo de doenças auto-imunes raras e por vezes graves, que incluem doenças pênfigoides e pemfigoides, epidermólise bullosa acquisita e dermatite herpetiforme Duhring. Uma característica comum das dermatoses auto-imunes bolhosas – com excepção da doença de Duhring – são auto-anticorpos dirigidos contra as proteínas estruturais da pele e das mucosas e responsáveis por uma perda de integridade cutânea [1].
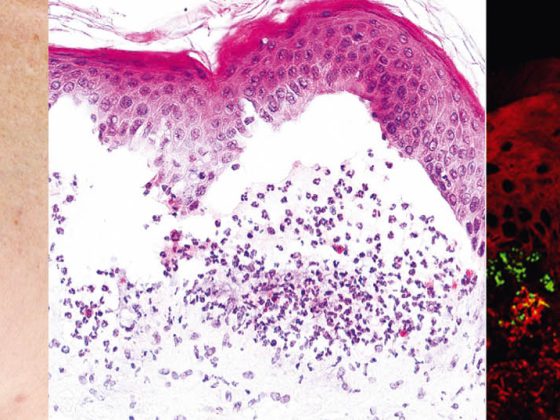
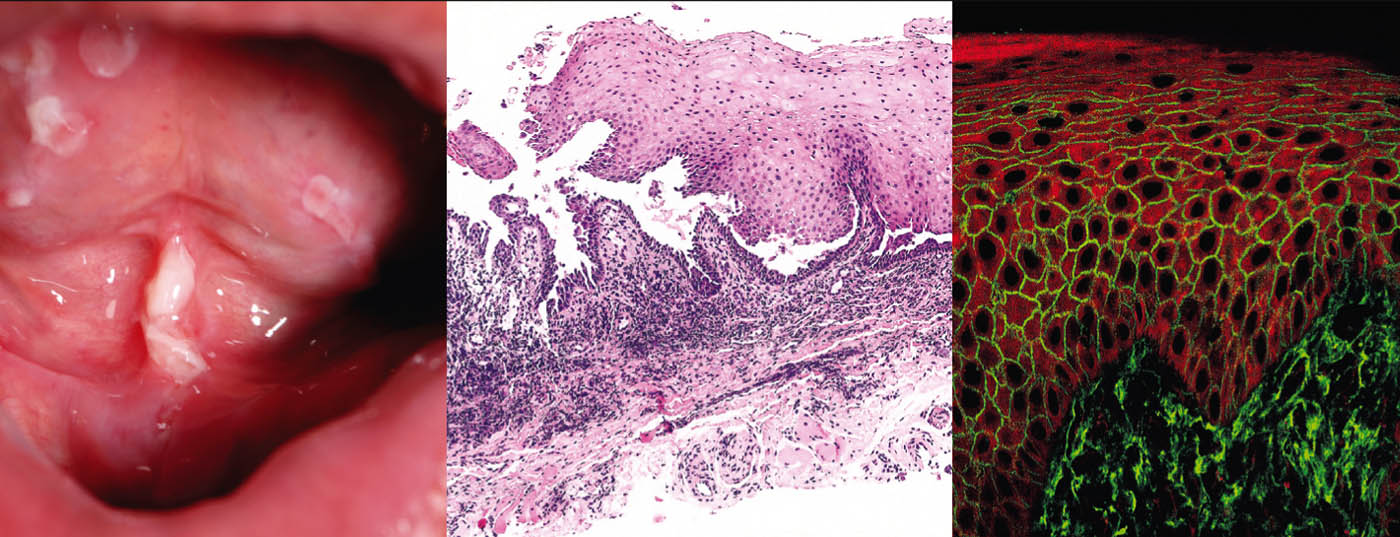
Pemphigus vulgaris (Tab. 1, Fig. 1) manifesta-se com bolhas generalizadas, mucocutâneas e flácidas que se rompem muito rapidamente, de modo que o quadro clínico é normalmente dominado por erosões e crostas.

O envolvimento das mucosas, especialmente na boca, que é muito doloroso, ocorre na maioria dos pacientes e por isso o diagnóstico não é raramente feito inicialmente pelos dentistas. Pathogeneticamente, o pemphigus vulgaris é caracterizado por autoanticorpos (AK) contra proteínas de adesão celular – desmogleins 1 e 3. Em pacientes com infestação exclusivamente oral, as AK são dirigidas contra a desmoglein 3. Histopatologicamente, há formação de fendas intra-epidérmicas suprabasais com acantólise e “formação de pedra tumular” dos queratinócitos basais. A imunofluorescência directa (DIF) mostra depósitos típicos de IgG e C3 intercelulares na epiderme. A imunofluorescência indirecta (IIF) do soro do doente confirma a circulação de auto-anticorpos IgG. Usando ELISA, a detecção directa de desmoglein 3 e desmoglein 1 pode ser conseguida no soro do paciente [2].
A terapia destina-se principalmente a reduzir a produção de auto-anticorpos. Corticosteróides sistémicos e outros imunossupressores tais como azatioprina, micofenolato mofetil, ciclofosfamida ou ciclosporina continuam a ser as terapias padrão. Outras opções são a plasmaférese ou imunoglobulinas intravenosas (IVIG). Em casos resistentes à terapia, o rituximab anticorpo anti-CD20 em particular é uma alternativa promissora. Para além do pemphigus vulgaris, o grupo das doenças do pênfigo inclui também o pemphigus foliaceus [3], pemphigus herpetiformis [4], pemphigus paraneoplastic pemphigus [5] e IgA pemphigus.
Penfigóide Bolhoso
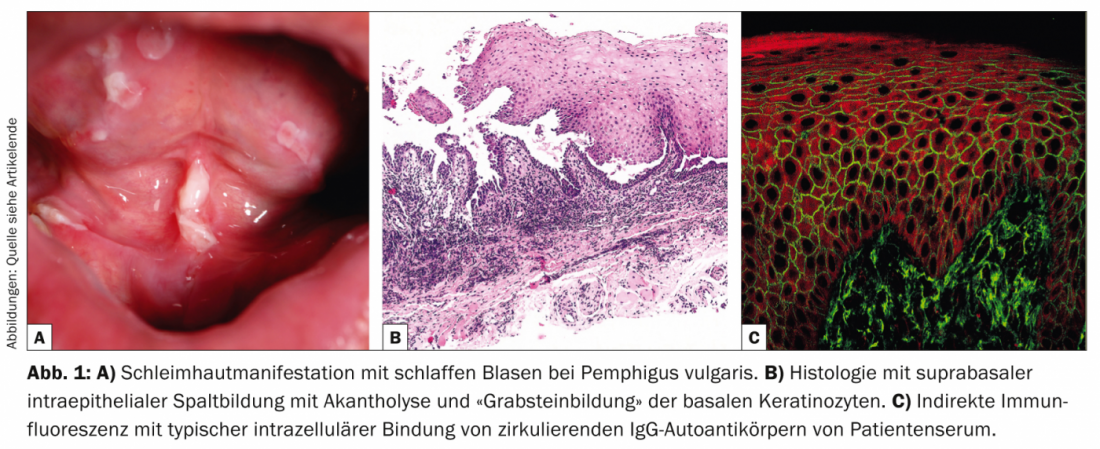
Com uma incidência de 12,1 novos casos/milhões/ano na Suíça, o pemfigoide bolhoso (Tab. 2, Fig. 2) é a doença mais frequente do grupo do pemfigoide e, ao mesmo tempo, a dermatose auto-imune formadora de bolhas mais frequente de todas [6].

O pemfigoide bolhoso ocorre geralmente na velhice e caracteriza-se por bolhas salientes em pele inflamada ou normal que provocam comichão intensa. Muitas vezes, esta doença funciona inicialmente sem bolhas e é diagnosticada como eczema, urticária ou prurigo devido ao prurido pronunciado. Patogenicamente, a doença é causada por auto-anticorpos contra a BP 180 (também conhecida como colagénio tipo XVII). A histologia mostra bolhas subepidérmicas com epiderme intacta e infiltrados proeminentes ricos em eosinófilos. Na DIF de pele perilesional, são encontrados depósitos de IgG ao longo da zona de junção dérmico-epidérmica (membrana do porão), que pode ser confirmada na IIF com a detecção de anticorpos IgG ligados à membrana do porão. O nível sérico de auto-anticorpos contra a BP180 correlaciona-se com a actividade da doença e pode ser determinado durante o curso para determinar a necessidade de mais terapia.
Os corticosteróides tópicos ou sistémicos continuam a ser a base da terapia. Outras opções incluem tetraciclinas com nicotinamidas, dapsona ou, em doenças graves, imunossupressores, especialmente azatioprina, bem como IVIG e rituximab em casos resistentes a terapias.
IgA linear, dermatose bolhosa
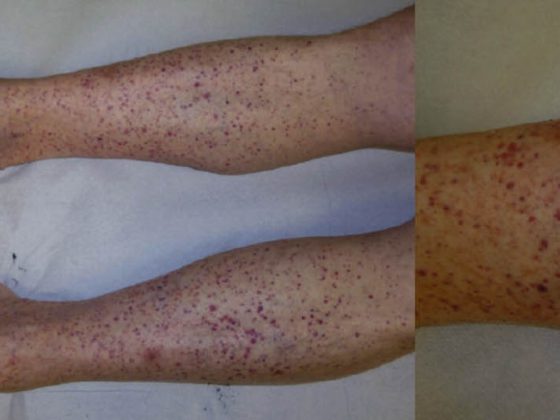
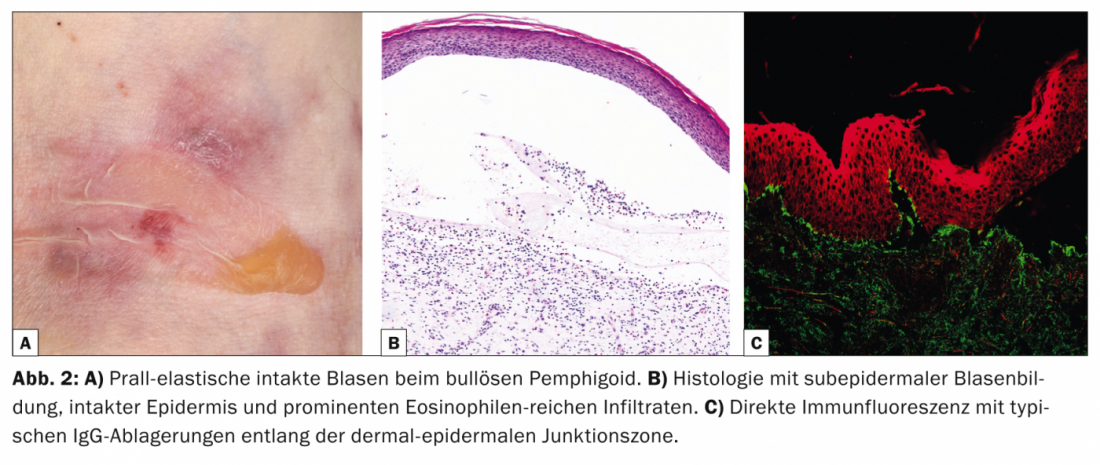
A dermatose linear IgA (LABD, Tab. 3, Fig. 3) caracteriza-se por vesículas e bulas pruriginosas, generalizadas e acentuadas por extensor. A doença ocorre tanto em adultos como em crianças.

Na infância, é a dermatose auto-imune bolhosa mais comum e é normalmente auto-limitada [7]; nos adultos, o LABD é frequentemente associado a drogas (vancomicina) [8]. Histologicamente, o LABD é semelhante à doença de Duhring na medida em que se caracteriza por bolhas subepidérmicas, epiderme intacta e infiltrações ricas em neutrófilos na junção dérmica-epidérmica. O DIF mostra depósitos lineares de IgA na área da zona da membrana do porão.
A IIF pode confirmar o diagnóstico através da detecção de auto-anticorpos IgA circulantes que se ligam ao telhado da bexiga. A maioria dos pacientes responde à terapia com dapsona ou sulfapiridinas. Também foram descritos tratamentos bem sucedidos em crianças e adultos com vários antibióticos (cicloxacilina, eritromicina, tetraciclina ou trimetoprim-sulfamethoxazol).
Epidermiólise bullosa aquisita
Epidermiolysis bullosa aquisita (EBA, Tab. 4, Fig. 4) está clinicamente dividida numa forma mecanobolhosa cicatrizada, não-inflamatória, localizada e muitas vezes acentuada, e numa variante generalizada, inflamatória e não cicatrizada. A doença ocorre principalmente na meia-idade adulta e é muito rara em crianças.

Existe uma estreita associação com a doença inflamatória intestinal [9]. Patogenicamente, a EBA caracteriza-se pela deposição de autoanticorpos IgG contra o procollagen de tipo VII do fibrilho de ancoragem. Histologicamente, o espécime de rotina mostra uma bolha subepidérmica com a epiderme intacta. Na DIF de pele perilesional, os anticorpos IgG são encontrados em bandas ao longo da zona de junção dermo-epidérmica. Na IIF sobre a pele humana separada por NaCl-, que é positiva em 50% dos casos, os IgG circulantes ou, menos frequentemente, os auto-anticorpos IgA ligam-se no chão da bexiga.
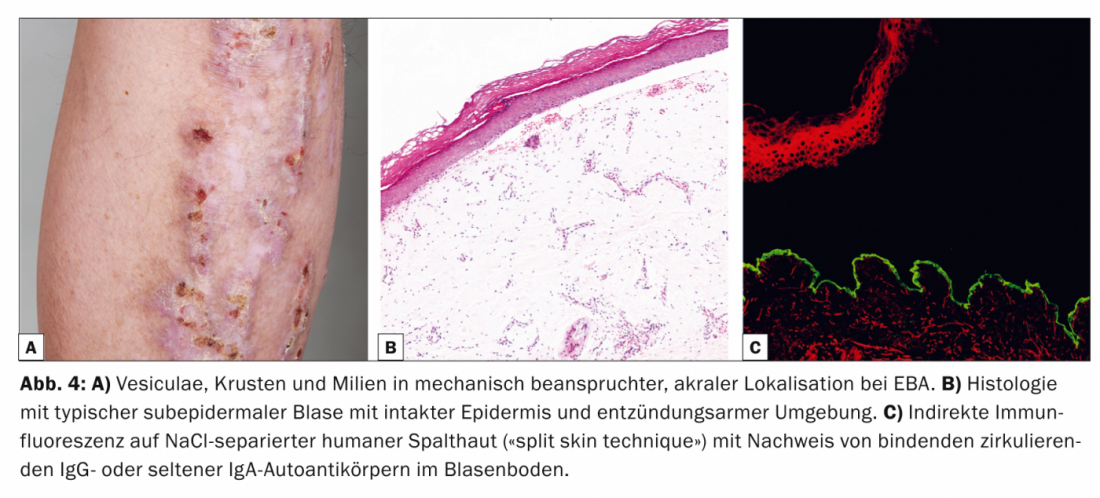
Western blot e ELISA podem ser utilizados complementarmente para detectar auto-anticorpos IgG circulantes [10]. A terapia dos MSDs é difícil, muitas vezes insatisfatória e visa principalmente um efeito imunossupressor. As opções de tratamento actuais são, portanto, principalmente corticosteróides sistémicos, dapsona ou colchicina. Além disso, há relatos de uma resposta positiva ao rituximab anticorpo anti-CD20 em casos resistentes ao tratamento.
Dermatite herpetiforme
Dermatite herpetiforme (DH, Tab. 5, Fig. 5) é uma rara manifestação cutânea de enteropatia sensível ao glúten (doença celíaca) e apresenta uma intensa comichão eritematosa e papulovesículas, que ocorrem principalmente no lado extensor e no sacro.

Patogenicamente, tanto a DH como a doença celíaca estão associadas ao genótipo HLA-DQ2, no qual estão presentes autoanticorpos IgA contra a reticulina, endomísio e transglutaminase tecidual, respectivamente. transglutaminase epidérmica. Histologicamente, há formação de fendas subepidérmicas e infiltrações ricas em neutrófilos com formação parcial de microabscess [11].
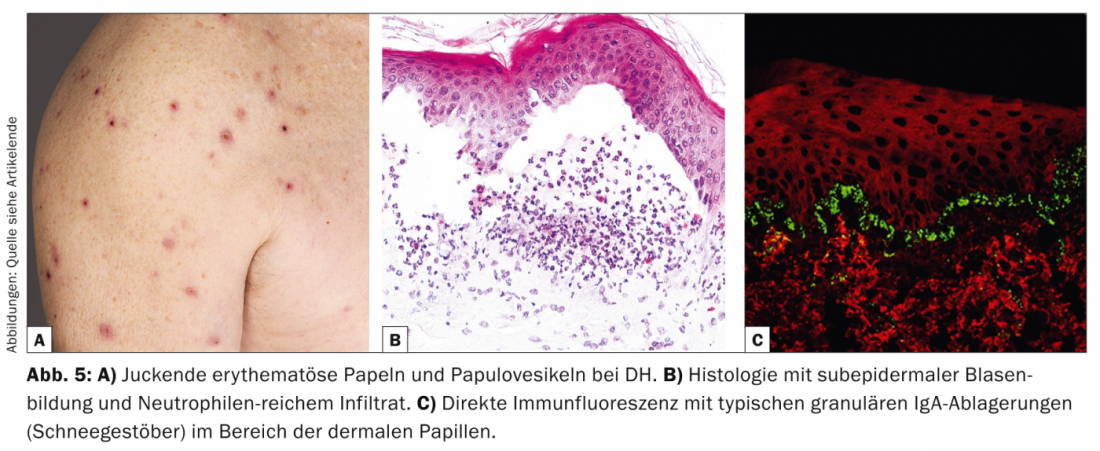
Histologicamente, a DH aparece semelhante à dermatose bula linear de IgA, mas em DIF de pele perilesional, depósitos granulares de IgA (flocos de neve) são encontrados na área das papilas dérmicas. Terapêuticamente, a dieta sem glúten está em primeiro plano, o que melhora tanto as queixas gastroenterológicas como os sintomas cutâneos [12]. As opções alternativas são dapsona, sulfapiridinas ou corticosteróides sistémicos. Dapsona melhora as lesões cutâneas mas não os sintomas intestinais.
Dra. Emmanuella Guenova, MD
Fontes de imagem:
Imagens clínicas: Arquivo fotográfico do Hospital Universitário de Zurique
Imagens histológicas: Dra. Emmanuella Guenova, MD
Ilustrações de imunofluorescência: Birgit Fehrenbacher
Literatura:
- Bolognia, JL, Jorizzo JL, Schaffer JV: Dermatologia. 2012: Elsevier Health Sciences UK.
- Chan LS: Doenças de pele com bolhas. 2009: Taylor & Francis.
- Guenova E, et al: Tinea incognito escondida sob o pênfigo foliáceo aparentemente resistente ao tratamento. Acta Derm Venereol 2008; 88(3): 276-277.
- Lebeau S, et al: Pemphigus herpetiformis: análise do perfil do autoanticorpo durante o curso da doença com alterações no fenótipo clínico. Clin Exp Dermatol 2010; 35(4): 366-372.
- Heizmann M, et al: Tratamento bem sucedido do pênfigo paraneoplásico em NHL folicular com rituximab: relatório de um caso e revisão do tratamento do pênfigo paraneoplásico em NHL e CLL. Am J Hematol 2001; 66(2): 142-144.
- Marazza G, et al: Incidência de pemfigoide bolhoso e pênfigo na Suíça: um estudo prospectivo de 2 anos. Br J Dermatol 2009; 161(4): 861-868.
- de las Heras MN: Dermatose bula linear IgA da infância: boa resposta ao tratamento antibiótico. Clin Exp Dermatol 2014; 39(3): 395-397.
- Tashima S, et al: Um caso de dermatose bullous IgA linear induzida por vancomicina com anticorpos IgA circulantes para o domínio NC16a de BP180. Int J Dermatol 2014; 53(3): 207-209.
- Hundorfean G, Neurfean MF, Sitaru C: Autoimunidade contra colagénio tipo VII na doença inflamatória intestinal. J Cell Mol Med 2010; 14(10): 2393-2403.
- Calabresi V, et al: Sensibilidade de diferentes ensaios para o diagnóstico serológico de epidermólise bullosa acquisita: análise de uma coorte de 24 pacientes italianos. J Eur Acad Dermatol Venereol 2014; 28(4): 483-490.
- Hall MA, Lanchbury JS, Ciclitira PJ: genes da região HLA classe II e susceptibilidade à dermatite herpetiforme: as associações DPB1 e TAP2 são secundárias às da subregião DQ. Eur J Immunogenet 1996; 23(4): 285-296.
- Hervonen K, et al: Dermatitis herpetiformis in children: um estudo de acompanhamento a longo prazo. Br J Dermatol 2014 [Epub ahead of print].
CONCLUSÃO PARA A PRÁTICA
- Em todas as dermatoses auto-imunes bolhosas, para além da história médica, o exame de todo o tegumento, incluindo a pele, é importante. a inspecção das mucosas e dos pregos é obrigatória.
- Se uma doença auto-imune bolhosa for clinicamente ou histologicamente suspeita, o diagnóstico deve ser confirmado pela detecção dos auto-anticorpos subjacentes (coloração dos anticorpos ligados aos tecidos em imunofluorescência directa na secção dos tecidos ou detecção no soro por imunofluorescência indirecta ou ELISA).
- Na terapia das dermatoses auto-imunes bolhosas, os imunossupressores clássicos ainda são utilizados (por exemplo, corticosteróides, azatioprina).
- As novas opções terapêuticas são anticorpos anti-CD20 (Rituximab), que conduzem a uma redução dos autoanticorpos.
PRÁTICA DA DERMATOLOGIA 2014; 24(4): 6-10