Os defeitos congénitos do coração são a malformação de órgãos mais comum nas crianças. Cada deficiência cardíaca requer uma avaliação e classificação cardiológica pediátrica. O acompanhamento cardiológico pediátrico especializado é essencial tendo em vista o crescimento da criança, possíveis descobertas cardíacas residuais após intervenção/cirurgia e quaisquer complicações tardias ao longo da infância e adolescência.
Os defeitos congénitos do coração são a malformação mais comum dos órgãos, ocorrendo em cerca de 1% de todos os recém-nascidos. Para a Suíça, isto correspondeu a cerca de 850 crianças com 85 287 nascidos vivos em 2014. O espectro varia desde simples defeitos cardíacos que têm pouco efeito no sistema cardiovascular até aos graves (Tab. 1) que podem levar à morte prematura se não forem tratados [1].

A frequência com que os defeitos cardíacos individuais ocorrem varia muito (Tab. 2) e cada defeito cardíaco requer uma avaliação e classificação cardiológica pediátrica. O método de exame diagnóstico mais importante é – para além do exame clínico, incluindo o exame do paciente – o exame do paciente. Auscultação – ecocardiografia, que permite a visualização e definição de praticamente todas as estruturas cardíacas.

O electrocardiograma (ECG) (também ECG Holter 24h) detecta quaisquer arritmias ou anomalias de condução, assim como sinais de hipertrofia. Dependendo da situação, são indicados outros métodos de exame especiais, tais como espiroergometria, ressonância magnética cardíaca (RMC), tomografia computorizada (TC) ou cateterização cardíaca para diagnóstico invasivo (por exemplo, medição de pressão, angiografias). Este último oferece também a possibilidade de intervenção ao mesmo tempo. Isto significa que hoje em dia a maioria dos defeitos cardíacos são diagnosticados no primeiro ano de vida. Sem terapia cirúrgica ou interventiva, a esperança de vida é significativamente reduzida para defeitos cardíacos moderados e graves.
Os extraordinários progressos feitos em cirurgia cardíaca, cardiologia interventiva, anestesia e medicina intensiva nas últimas décadas melhoraram significativamente as hipóteses de sobrevivência após a cirurgia cardíaca na infância. Por exemplo, mais de 90% das crianças com cardiopatias congénitas atingem agora a idade adulta [2,3]. Contudo, são frequentemente confrontados com problemas multifacetados no decurso das suas vidas devido à sua patofisiologia cardíaca e às doenças concomitantes que frequentemente também estão presentes. Um ponto chave é que a maioria dos defeitos cardíacos podem ser reparados mas não corrigidos no verdadeiro sentido [4].
Descobertas cardíacas residuais significativas e o crescimento da criança tornam essencial o acompanhamento especializado em cardiologia pediátrica. Um acompanhamento óptimo das crianças com doenças cardíacas requer uma estreita colaboração entre médicos de clínica geral e pediatras no consultório privado e os médicos especialistas envolvidos. Isto é particularmente importante no caso de doenças complexas concomitantes que prejudicam o desenvolvimento neurológico, por exemplo [5].
Na adolescência, a transição para a medicina adulta para um cardiologista especificamente formado (cardiologista GUCH; GUCH = Grown-Ups with Congenital Heart Disease) deve ser iniciada a tempo no sentido de um fluxo contínuo de informação [6].
Complicações no curso
Arritmias: Alguns defeitos cardíacos estão associados a arritmias precoces. Por exemplo, o bloqueio atrioventricular completo pode ocorrer com a transposição cc das grandes artérias. No entanto, a maioria das arritmias ocorre devido a resíduos hemodinâmicos, tais como carga de pressão persistente com fibrose ventricular ou carga de volume e/ou cicatrização miocárdica (atrio-ventricotomia), após cirurgia cardíaca. Uma desordem do ritmo, incl. Transtorno de condução, pode desenvolver-se cedo ou tarde no pós-operatório. As observações incluem taquicardia supraventricular (geralmente taquicardia de reentrada), flutter ou fibrilação atrial, taquicardia atrial ectópica, síndrome do seio doente, taquicardia ventricular, bloqueio atrioventricular e morte súbita cardíaca.
Enquanto o risco de arritmia permanece baixo após operações cardíacas mais simples, aumenta significativamente após operações complexas como a paliação de Fontan em corações univentriculares. Por esta razão, o ECG e a ergometria (a longo prazo) fazem parte da avaliação médica regular destes pacientes. Para as arritmias que ocorrem tardiamente no pós-operatório, a electrofisiologia invasiva está hoje disponível, além da terapia medicamentosa, que alcança bons resultados através da ablação de, por exemplo, cicatrizes intracardíacas.
Limitações físicas: Com o número crescente de crianças que chegam à idade adulta após reparação cirúrgica ou paliação, a inclusão de crianças no tecido social com os seus pares tornou-se um objectivo importante. Enquanto um paciente saudável pode quintuplicar o seu débito cardíaco durante o exercício, um paciente com um defeito cardíaco complexo, tal como após paliação de Fontan no coração univentricular, é na melhor das hipóteses capaz de o duplicar. Isto resulta numa resiliência física limitada [7].
A actividade desportiva na área do enduro é geralmente apoiada. Em contraste, o treino de pesos isométricos ou desportos de competição não são recomendados para a maioria dos pacientes.
Os testes e a avaliação do desempenho físico através da espiroergometria são uma componente regular dos exames de acompanhamento cardiológico pediátrico a partir da idade de cerca de dez anos.
Fracasso no desenvolvimento/neurocognitivo: Crianças com um defeito cardíaco grave podem desenvolver um fracasso no desenvolvimento e são então mais pequenas e mais magras em estatura do que as crianças saudáveis da mesma idade. Vários estudos demonstraram que crianças com defeitos cardíacos complexos têm um risco acrescido de défices neurocognitivos após a cirurgia cardíaca em neonatologia ou na infância. Atraso no desenvolvimento de capacidades motoras finas e brutas, problemas comportamentais, atenção reduzida, sintomas de hiperactividade e atraso linguístico são défices comuns [6,8]. Isto pode ter um impacto negativo na vida quotidiana e nas carreiras educacionais e profissionais na idade adulta. As crianças com defeitos cardíacos ligeiros a moderados não são, na sua maioria, afectadas. É importante levantar estas questões com os pais e oferecer apoio direccionado ao pediatra supervisor ou ao médico de família.
Endocardite: A endocardite infecciosa é uma complicação grave e potencialmente fatal em doentes com doença cardíaca congénita ou doença cardíaca adquirida. É por isso que a educação dos pais e dos pacientes é de grande importância durante os exames cardiológicos pediátricos. Os sintomas de endocardite devem ser reconhecidos precocemente para que se possa dar uma resposta adequada. Os benefícios e a correcta administração da profilaxia de endocardite com medicamentos também devem ser comunicados. As actuais directrizes para a profilaxia da endocardite em crianças na Suíça estão listadas no quadro 3.

Hipertensão arterial pulmonar: Todos os defeitos cardíacos congénitos com grandes shunts intra ou extracardíacos levam a uma carga de volume e pressão ilimitada do leito vascular pulmonar. Como resultado, a hipertensão arterial pulmonar pode desenvolver-se, levando a alterações irreversíveis nas fases finais desta grave doença. Por exemplo, no caso de um grande defeito do septo ventricular não corrigido com shunt esquerda-direita, há um aumento da pressão no leito vascular pulmonar ao longo dos anos, que termina numa inversão do shunt com cianose devido a um novo shunt direita-esquerda (reacção Eisenmenger). Um dos objectivos da intervenção cirúrgica na primeira infância é evitar este desenvolvimento. Os pacientes com defeitos cardíacos congénitos têm um risco aumentado de desenvolver hipertensão arterial pulmonar mesmo sem a presença de shunts relevantes. Dependendo da sua severidade, isto tem um efeito prejudicial na qualidade e duração da vida [10]. Para o diagnóstico e início de uma terapia específica, é geralmente necessário estar ligado a um centro especializado.
Doenças concomitantes: Os defeitos congénitos do coração estão associados a outras malformações e/ou anomalias cromossómicas em 15-20%. Crianças com trissomia do cromossomo 21 têm vitium cordis em 40-50% dos casos. Estes pacientes, muitas vezes com malformações complexas, beneficiam de cuidados multidisciplinares.
Exemplos de cuidados de acompanhamento e aspectos específicos de defeitos cardíacos individuais
Estenose do istmo aórtico (AIST): O objectivo é eliminar a estenose numa fase inicial e criar uma aorta tão livre de gradientes e de calibre normal quanto possível. Isto só pode ser conseguido por cirurgia ou por cateterização cardíaca interventiva (dilatação de balão ou implante de stent) (Fig. 1). O método de tratamento depende da idade do paciente, bem como da morfologia do AIST. Existem frequentemente malformações associadas (por exemplo, válvula aórtica bicúspide em cerca de 50% dos casos).
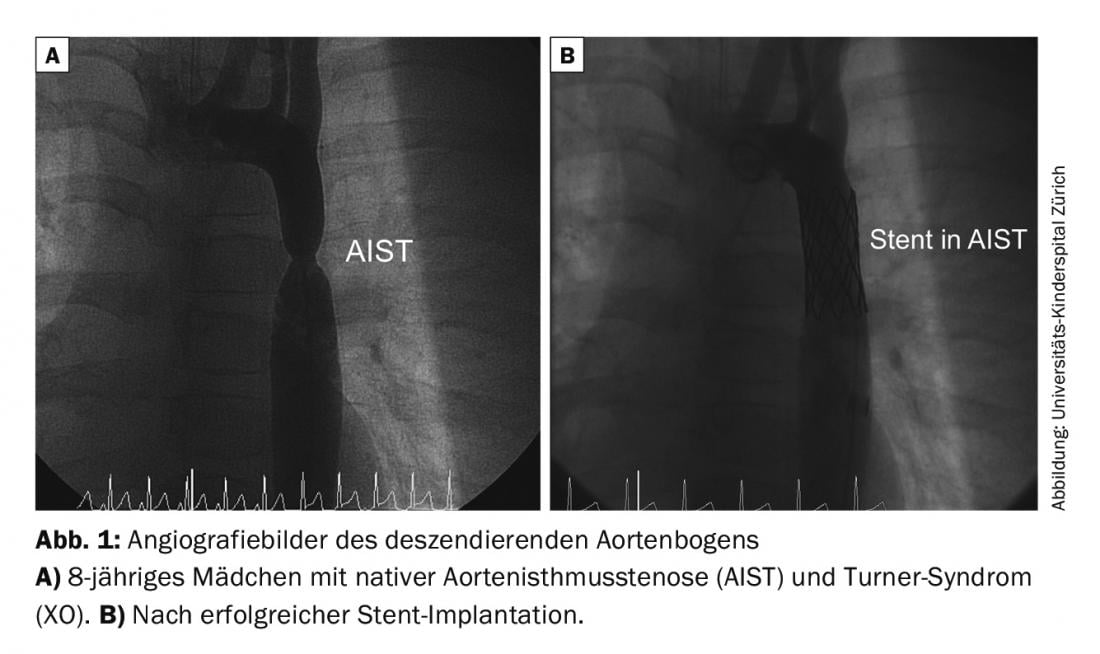
Os pacientes com reparação cirúrgica têm um risco de 5-10% de reestenose no curso. O risco de hipertensão arterial aumenta com a idade, mesmo que a estenose tenha sido efectivamente removida. Outros riscos possíveis são o desenvolvimento de aneurismas/dissecções da aorta no local da sutura ou no stent.
Recomendações de acompanhamento [11]:
- Normalmente anualmente, ao longo de pelo menos um ano. de 2 em 2 anos:
- estado clínico
- Medição da pressão arterial (PA) em todas as 4 extremidades
- dependendo da PA em repouso: 24h de medição da PA
- ECG
- Ecocardiografia (tamanho do ventrículo esquerdo, espessura e função do miocárdio, grau de estenose sobre o arco aórtico/stmo, prestar mais atenção às malformações associadas, se necessário)
- Ergometria (a partir de 10 anos) a cada 3-4 anos
- cMRI: em caso de visualização insuficiente na ecocardiografia: visualização do arco aórtico, exclusão da formação do aneurisma de 5 em 5 anos
d-transposição das grandes artérias (d-TGA): Actualmente, a cirurgia de troca arterial para restabelecer a concordância ventrículo-arterial da d-TGA é o tratamento de primeira escolha. A aorta e a artéria pulmonar são intercambiadas e através da chamada manobra de Lecompte, a bifurcação pulmonar encontra-se finalmente em frente da aorta (Fig. 2) . Ao mesmo tempo, é necessária a reimplantação das artérias coronárias. A válvula pulmonar anatómica serve assim como uma válvula neo-aórtica na circulação sistémica para toda a vida.
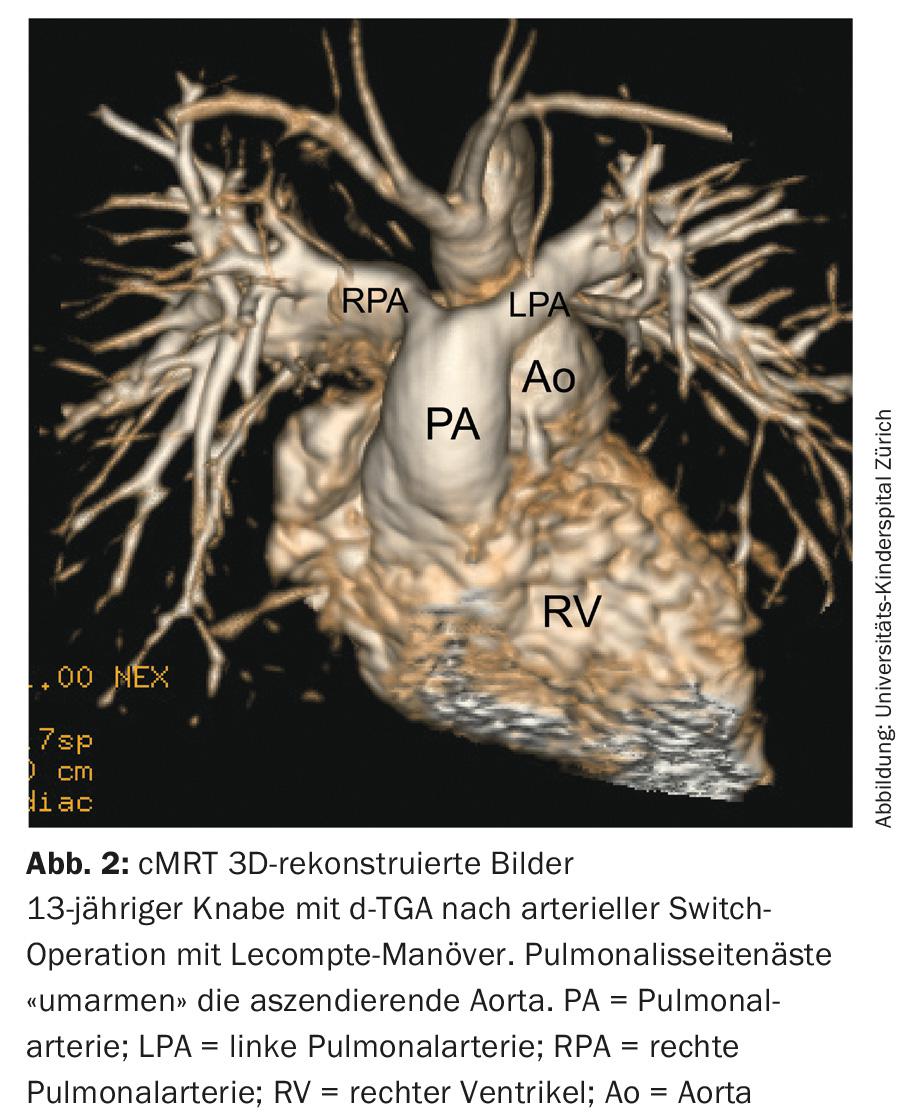
A longo prazo, esta válvula neo-aórtica pode tornar-se disfuncional e a raiz aórtica pode dilatar. Devido à manobra lecompte com deslocamento dos ramos colaterais pulmonares anterior à aorta ascendente, pode ocorrer torção e alongamento e assim estenose dos ramos colaterais pulmonares. Como a operação envolve transferência coronária, a torção e alongamento também pode levar à estenose ou, em casos extremos, à oclusão de uma artéria coronária, com enfarte consecutivo do miocárdio e morte cardíaca súbita.
Recomendações de acompanhamento [9,11]:
- Normalmente anualmente, ao longo de pelo menos um ano. de 2 em 2 anos:
- estado clínico
- ECG
- Ecocardiografia (tamanho e função ventricular, grau de estenose dos ramos laterais pulmonares, distúrbio de mobilidade da parede, função da válvula neo aórtica, dimensão da raiz neo aórtica).
- ECG a longo prazo de acordo com a avaliação individual
- Angiografia coronária em curso perioperatório complicado, doente sintomático ou diagnóstico de rotina anormal.
- Ergometria (a partir de 10 anos) a cada 2-3 anos
- cMRI: em caso de má qualidade sonora, para avaliação do tamanho e função dos ventrículos direito e esquerdo e avaliação dos ramos laterais pulmonares. Avaliação das artérias coronárias e/ou cicatrizes do miocárdio.
Tetralogia de Fallot (TOF): O objectivo da reparação cirúrgica anatómica da tetralogia de Fallot é o fecho de retalho VSD e a eliminação da obstrução da via de saída do ventrículo direito (RVOTO). Se o anel valvar pulmonar for pequeno em diâmetro e/ou a válvula for displásica, é necessário o implante de uma placa transannular, o que leva sempre à insuficiência valvar pulmonar (Fig. 3) . A médio e longo prazo, isto resulta numa dilatação ventricular direita relevante, que pode levar a disfunção ventricular, desempenho prejudicado e arritmia e, em última análise, requer a substituição da valva pulmonar.

O bloqueio completo do ramo direito é comum após o fecho do penso VSD; o risco de bloqueio atrioventricular completo que requer a implantação de um pacemaker é pequeno (<2%).
Recomendações de acompanhamento [9,11]:
- Normalmente anualmente
- estado clínico
- ECG (QRS largura)
- Ecocardiografia (RVOTO residual incluindo ramos laterais pulmonares, regurgitação valvar pulmonar, tamanho e função ventricular direita, CIV residual, tamanho e função ventricular esquerda, dimensão da raiz aórtica).
- ECG a longo prazo (taquicardia não sustentada) min. de 3 em 3 anos em doentes assintomáticos
- Ergometria (a partir de 10 anos), a cada 3-5 anos
- cMRI: com dilatação crescente do VD: para avaliação do tamanho e função do ventrículo direito e avaliação da insuficiência pulmonar (fracção regurgitante), tamanho e função ventricular esquerda. Avaliação dos ramos laterais pulmonares.
Ventrículo único (VS): No caso de corações univentriculares, é feita uma distinção básica entre duas formas básicas, dependendo se o ventrículo direito ou esquerdo não se formou suficientemente (por exemplo, atresia tricúspide ou síndrome do coração esquerdo hipoplásico); Fig. 4). A terapia cirúrgica sob a forma de três intervenções paliativas geralmente escalonadas termina na “circulação de Fontan”. O objectivo é criar um circuito ligado em série a partir dos circuitos paralelos que têm existido até agora. No período neonatal, o primeiro passo é estabilizar a circulação paralela, que, dependendo do defeito cardíaco, envolve geralmente um shunt ou uma redução da hiperperfusão pulmonar através de uma banda. Num segundo passo, uma anastomose cavopulmonar superior bidireccional (Glenn) é criada na infância, resultando numa descompressão significativa do volume do coração. Com a conclusão de Fontan aos 2-3 anos de idade, utilizando um conduto extracardíaco, anastomose cavopulmonar total, consegue-se uma paliação definitiva em doentes com corações univentriculares. Apenas com esta separação final da circulação é levantada a cianose (para além de pequenos shunts residuais; colaterais ou fenestração).
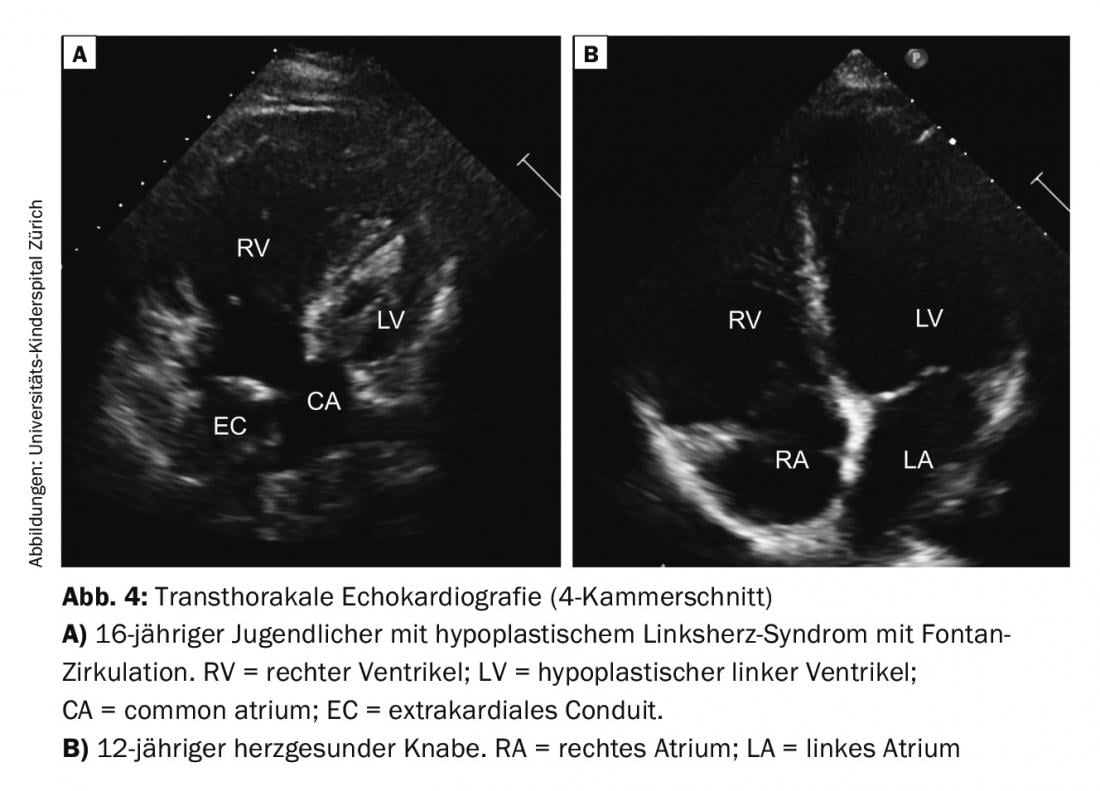
O cuidado durante as etapas cirúrgicas individuais e o acompanhamento deste grupo heterogéneo e único de pacientes com todas as suas complicações e efeitos tardios é um grande desafio para os cardiologistas pediátricos e mais tarde para os cardiologistas adultos especializados (cardiologista GUCH).
As complicações nestes pacientes são muito diversas: função ventricular reduzida, insuficiência valvar AV, estenose na circulação sistémica ou pulmonar, arritmia, incompetência cronotrópica, trombose/embolismo, enteropatia por perda de proteínas, bronquite plástica, disfunção hepática, hipertensão pulmonar, paresia diafragmática, escoliose, etc.
Recomendações para o seguimento após a conclusão da fontan [9,11]:
- min. anual
- estado clínico
- Medição da PA em todas as 4 extremidades
- Saturação transcutânea de oxigénio
- ECG
- Ecocardiografia (função SV, insuficiência da válvula AV, obstrução esquerda (estenose subaórtica, estenose istómica), perfil de fluxo da anastomose cavopulmonar superior ou na circulação de Fontan e veias hepáticas, etc.)
- Exame laboratorial: proteínas totais ou albumina sérica, valores hepáticos, pró-BNP como parâmetro de função cardíaca, hemoglobina.
- ECG a longo prazo (taquicardia reentrante intra-atrial não sustentada, competência cronotrópica), se houver indícios de arritmia ou pelo menos alguns sinais de um ataque cardíaco. todos
- 3 anos
- Ergometria (a partir de 10 anos), mínimo. De 3 em 3 anos
- cMRI: Avaliação da função ventricular, quantificação do fluxo sanguíneo, visualização anatómica das ligações de Fontan. A implementação depende do curso clínico, mas é geralmente recomendada na adolescência e antes da transição para os cardiologistas GUCH.
- Cateterização cardíaca diagnóstica: em caso de deterioração da circulação de Fontan, tais como diminuição da capacidade de exercício, aumento da cianose, ascites ou enteropatia por perda de proteínas, bronquite plástica, etc.
Palavras de encerramento
Actualmente, a maioria das cardiopatias congénitas têm um bom prognóstico a longo prazo. A detecção precoce do defeito cardíaco, o tratamento cirúrgico e/ou intervencionista por uma equipa experiente e cuidados de acompanhamento cuidadosos contribuem decisivamente para isso. A estreita cooperação entre pediatras e cardiologistas pediátricos, bem como outros especialistas, se necessário, e a posterior transição para um cardiologista GUCH é essencial.
Literatura:
- Hoffmann JIE, et al: The Incidence of Congenital Heart Disease (Incidência de Doença Cardíaca Congénita). JACC 2002; 39(12): 1890-1900.
- Warnes CA, et al: Task Force 1: The Changing Profile of Congenital Heart Disease in Adult Life. JACC 2001; 37(5): 1161-1198.
- Moons P, et al: Temporal Trends in Survival to Adulthood Among Patients Born with Congenital Heart Disease from 1970 to 1991 in Belgium. Circulação 2010; 122: 2264-2272.
- Oechslin E: Defeitos cardíacos congénitos: cuidados especializados ao longo da vida para uma doença crónica. Medicina Cardiovascular 2006; 9: 373-375.
- Mackie A: Crianças e Adultos com Doença Cardíaca Congénita Perdidos para Acompanhamento. Circulação 2009; 120(4): 302-309.
- Bauersfeld U: Transição, transferência e cooperação em doentes com defeitos cardíacos congénitos – colaboração contínua de cardiologia pediátrica e de adultos. Medicina Cardiovascular 2006; 9: 336-341.
- Gewillig M, et al: Failure of the Fontan Circulation. Clínica de insuficiência cardíaca 2014; 10(1): 105-116.
- Sterken C, et al: Desenvolvimento Neurocognitivo após Cirurgia Cardíaca Pediátrica. Pediatria 2016; 137(6), doi: 10.1542/peds.2015-4675.
- Wernovsky G, et al: Guidelines for the Outpatient Management of Complex Congenital Heart Disease. Congenit Heart Dis 2006; 1: 10-26.
- Dimopoulos K, et al: Hipertensão pulmonar relacionada com doenças cardíacas congénitas: um apelo à acção. Eur Heart J 2014; 35(11): 691-700.
- Sociedade Alemã de Cardiologia Pediátrica e.V., ed. Schmaltz AA: Directrizes para diagnóstico e terapia em cardiologia pediátrica. Elsevier GmbH, Urban & Fischer Verlag 2007.
- Knirsch W, et al: Novas recomendações para a profilaxia de endocardite antibiótica em crianças na Suíça. Paediatrica 2009; 20(4): 28-34.
CARDIOVASC 2016; 15(5): 4-9