A hemoglobinúria paroxística nocturna (HNP) é uma doença adquirida rara, caracterizada por hemólise intravascular e hemoglobinúria. Devido a uma perda prolongada de hemoglobina através da urina, a PNH pode levar à deficiência de ferro e aumentar a suspeita de anemia. Contudo, a PNH é uma doença extremamente heterogénea com diversas manifestações e frequentemente sobrepõe-se a outras doenças da medula óssea.
A PNH é uma doença muito rara com uma incidência de cerca de 1,3/1.000.000 e uma prevalência de cerca de 16 por milhão de habitantes. Os sintomas foram descritos pela primeira vez como hemoglobinúria paroxística em 1882 pelo Dr. Strübing da policlínica médica de Greifswald. Marchiafava e Micheli introduziram o termo sindroma Marchiafava-Micheli, que ainda hoje é utilizado como sinónimo da doença. O nome PNH foi cunhado pelas três características desta doença: Paroxística= ocorre em episódios; Nocturna= ocorre à noite; Hemoglobinúria= hemoglobina na urina. Hoje em dia sabemos que a hemólise também pode ocorrer sem sintomas e é constante ao longo do dia. Além disso, apenas um em cada quatro pacientes tem hemoglobina na sua urina.
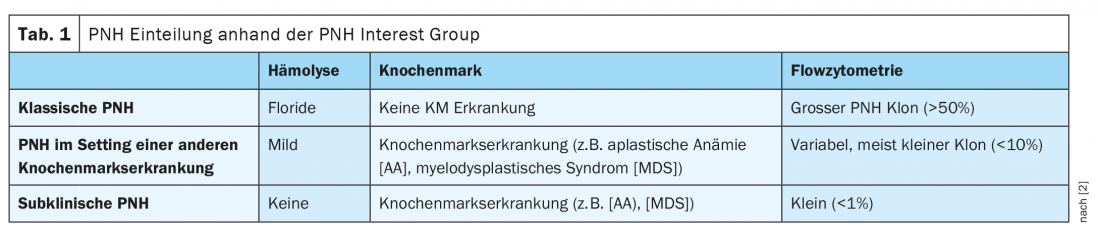
Pode ser dividido em três formas
A PNH pode ser dividida em três formas de acordo com a extensão da hemólise, os resultados da medula óssea e a citometria de fluxo. No PNH clássico, o doente tem hemólise muito marcada, mas nenhuma outra doença da medula óssea. Além disso, estes pacientes têm um grande clone PNH >50%. Para além da PNH clássica, a doença pode também ocorrer no contexto de outras doenças da medula óssea, classicamente em anemia aplástica. Estes pacientes não são geralmente sintomáticos e têm apenas hemólise ligeira e um pequeno clone variável PNH <10%. O PNH subclínico ocorre frequentemente em doenças da medula óssea, tais como anemia aplástica ou síndrome mielodisplásica. Normalmente, apenas um clone PNH muito pequeno é detectado nestes casos <1%, sem quaisquer sinais de hemólise (Tab. 1) [2]. Além disso, a PNH pode sobrepor-se a outras doenças em qualquer altura. É possível uma transição para a anemia aplástica (AA), bem como para a leucemia mielóide aguda (LMA) e síndrome mielodisplásica (MDS).
Começa sempre com PIGA
PNH é devido a uma mutação do gene para inositol inositol inositol A (PIGA) ligada ao x. Esta enzima está envolvida na biossíntese da chamada âncora GPI. A âncora GPI fixa certas proteínas à superfície da membrana celular. Estas proteínas ligadas incluem em particular o Factor Acelerador de Decaimento do Complemento (CD55) e o Inibidor de Lise Reactiva de Membrana (CD59). Estes dois componentes servem para defender contra o ataque complementar à membrana eritrócita. A ausência parcial ou total da âncora GPI e uma omissão correspondente das proteínas de superfície reguladora do complemento significa que a membrana eritrócita não está protegida de forma adequada. O resultado é uma hemólise intravascular. Entre outras coisas, a hemólise intravascular leva à formação de hemoglobina livre no sangue, que liga o óxido nítrico. O resultado é uma disfunção endotelial com contracção muscular lisa consecutiva e activação e agregação plaquetária, levando a um risco acrescido de tromboembolismo.
Vale a pena mencionar que os pacientes com uma mutação PIGA também têm normalmente outras mutações somáticas. Em hematologia, falamos então de alterações clonais ou clones. Esta é uma razão para diferentes cursos, e em alguns casos pode desenvolver-se uma doença maligna.
Triássico clássico
Os pacientes com PNH mostram uma tríade clássica constituída por hemólise, tromboembolismo e possível falha da medula óssea. Isto significa que podem ocorrer queixas diferentes em momentos diferentes. Estes incluem disfagia (41%), hipertensão pulmonar (47%), dispneia (66%), dor abdominal (59%), insuficiência renal (64%), disfunção eréctil (47%), hemoglobinúria (26%) e trombose (40%). No entanto, os principais sintomas incluem anemia (88%) e fadiga pronunciada (97%). Contudo, para além destes sintomas, também podem ocorrer apresentações atípicas, como um relatório de caso da Suíça demonstrou. Neste caso particular, o PNH foi eventualmente detectado num doente com enfarte do miocárdio, indicando trombose arterial [3]. Outra publicação do New England Journal, relata um jovem doente com dores de cabeça, dores abdominais, anemia e trombocitopenia, que teve trombose da veia sinusal como uma complicação da PNH [4]. Segundo a Dra. med. Beatrice Drexler do Hospital Universitário de Basileia, as tromboses são geralmente muito relevantes em doentes com PNH porque influenciam a mortalidade. Normalmente não afectam apenas as veias profundas das pernas, mas podem ocorrer em locais bastante atípicos [1].
Citometria de fluxo como padrão de ouro
O padrão de ouro actual para confirmação diagnóstica é a citometria de fluxo do sangue periférico, que detecta a falta de expressão de moléculas ancoradas em GPI em eritrócitos e leucócitos. Neste método, as células a examinar fluem uma após a outra através de uma fina câmara de medição, a chamada célula de fluxo. Certos antigénios de superfície das células são rotulados com anticorpos monoclonais fluorescentes a fim de se poder medir a fluorescência de cada partícula excitada pela luz laser. O critério de diagnóstico mínimo é uma população deficiente em GPI para pelo menos duas proteínas diferentes com uma cadeia de GPI em duas linhas celulares diferentes. Também relevante é o tipo e tamanho do clone PNH, que também pode ser definido em citometria de fluxo e fornecer informações sobre o risco de trombose. Indicações para citometria de fluxo são hemólise, trombose e falência da medula óssea (Fig. 1) [5,6].
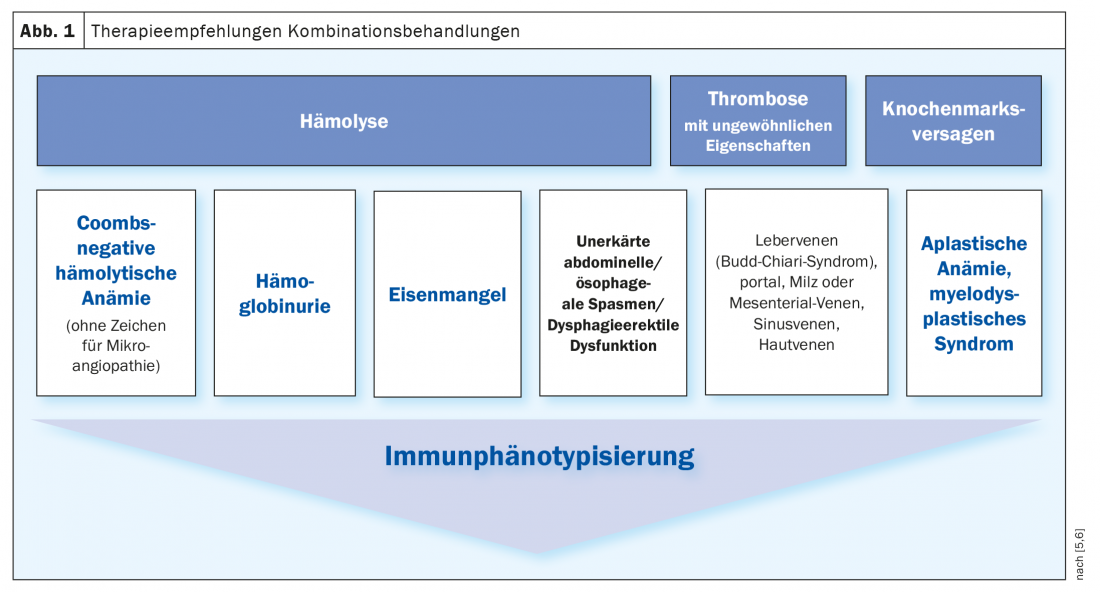
Algoritmo terapêutico
Em pacientes assintomáticos com valores laboratoriais pouco evidentes, não é normalmente necessário qualquer tratamento. No entanto, estes doentes devem ser acompanhados regularmente com contagens de sangue, parâmetros de hemólise e citometria de fluxo. Para os doentes que necessitam de tratamento, o foco é a terapia sintomática. A substituição do ácido fólico é obrigatória, bem como a administração de vitamina B12 e ferro, dependendo dos valores laboratoriais. Dependendo do tamanho do clone e conforme a necessidade, podem ser tomadas outras medidas de apoio. Além disso, a inibição da via do complemento terminal é feita por um anticorpo monoclonal como o eculizumab ou ravalizumab. Deve ser dada uma infusão com estes inibidores de elogios a cada duas ou oito semanas. Isto pode inibir a hemólise intravascular completa, reduzir a necessidade de transfusões, melhorar a qualidade de vida dos pacientes e também reduzir o risco de eventos tromboembólicos, que são a principal causa de morte na HNP.
Mensagens Take-Home
- A PNH é uma doença heterogénea com diversas manifestações.
- Muitas vezes sobrepõe-se a outras doenças da medula óssea (anemia aplástica e síndrome mielodisplásica).
- O padrão de ouro para o diagnóstico é a citometria de fluxo a partir do sangue periférico.
- O prognóstico melhorou significativamente com a administração de inibidores de complemento.
- No entanto, a terapia é muito dispendiosa.
- Outras terapias modificadoras de doenças estão em desenvolvimento.
Literatura:
- Dra. med. Beatrice Drexler, Hospital Universitário de Basileia, “Was wenn Eisen nicht wegen Eisen mangelt?”, conferência Iron Academy, 17.06.2021.
- Parker, et al: Diagnóstico e gestão da hemoglobinúria paroxística nocturna. Blood 2005, doi: 10.1182/blood-2005-04-1717.
- Gerber, et al: Inibição complementar para tratar o enfarte do miocárdio? BMJ Case Rep. 2011, doi: 10.1136/bcr.01.2011.3701.
- Sykes, et al: Case 40-2017: Uma mulher de 32 anos com dores de cabeça, dores abdominais, anemia e trombocitopenia. N Engl J Med. 2017, doi: 10.1056/NEJMcpc1710566.
- Röth, et al: Screening and Diagnostic Clinical Algorithm for Nocturnal Hemoglobinuria: Consenso de especialistas. Eur J Haematol 2018, doi: 10.1111/ejh.13059.
- Borowitz, et al: Cytometry B Clin Cytom 78:211-230. Drexler B Pipette – Swiss Laboratory Medicine 2010, No 1/2 March 2021.
PRÁTICA DO GP 2021; 16(9): 20-22