A síndrome de Lynch, também conhecida como HNPCC (cancro colorrectal hereditário não-polipose), é a causa hereditária mais comum de cancro colorrectal em todo o mundo. Além da cirurgia preventiva, a aspirina é também utilizada para a profilaxia do cancro. Quando deve pensar na síndrome de Lynch e o que deve fazer se suspeitar dela?
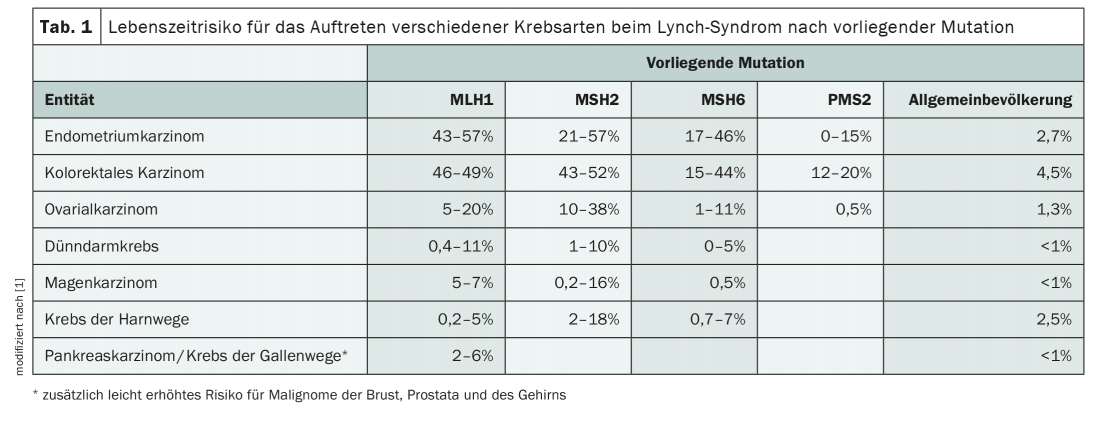
A síndrome do linchamento, herdada de forma autossómica dominante, aumenta substancialmente o risco de desenvolvimento de vários tipos de cancro. Para além dos carcinomas endometriais e colorrectais, outras doenças malignas tais como tumores ovarianos, pancreáticos ou gástricos também ocorrem frequentemente (Tab. 1) . Embora as mutações que compõem a síndrome do HNPCC sejam raras na população, com uma prevalência de 1:270 a 1:440, representam no entanto a predisposição hereditária mais frequente do cancro [1]. O diagnóstico precoce e as medidas preventivas que envolvem toda a família podem prevenir doenças malignas.
Reparação de ADN defeituoso
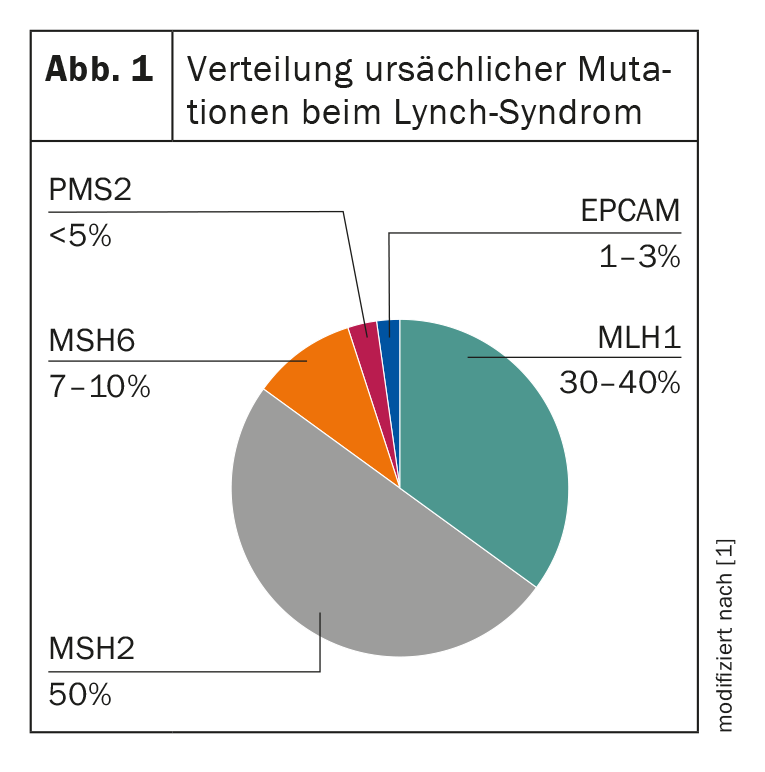
Nos indivíduos afectados, pode ser detectado um defeito genético na reparação do ADN, que se reflecte num alongamento de segmentos repetitivos de ADN – os chamados microssatélites. Estes diferem entre tecido tumoral e tecido saudável em pacientes com HNPCC, que é chamado de “instabilidade por microsatélite” (MSI). A presença de tal MSI com suspeita clínica simultânea é, portanto, altamente susceptível de indicar a síndrome de Lynch [2]. Até agora, são conhecidos quatro genes de reparação de incompatibilidade de DNA (MMR) cujas mutações na linha germinal conduzem ao desenvolvimento do HNPCC: MLH1, MSH2, MSH6 e PMS2. Além disso, a eliminação da linha germinal do gene EPCAM é uma causa possível, o que também resulta numa perda da proteína MSH2 no tumor (Fig. 1) . Uma perda de expressão correspondente nas células tumorais pode ser demonstrada pela imuno-histoquímica e o diagnóstico subsequentemente confirmado pela genética molecular [2]. A identificação da mutação familiar é de grande importância, uma vez que afecta não só o próprio paciente, mas também toda a sua família. Permite testar de forma preditiva os parentes saudáveis, que é o que torna as medidas preventivas possíveis em primeiro lugar. O risco de contrair a doença difere em função da mutação e da idade. Isto pode ser calculado individualmente utilizando a base de dados da futura síndrome de Lynch [3].
Devido à herança autossómica dominante, existe normalmente uma cópia defeituosa do gene desencadeador na linha germinal, que é transmitida com uma probabilidade de 50%. Se ocorrem mutações somáticas, isto é, aleatórias, na segunda cópia genética – originalmente funcional -, ocorre o defeito de reparação do ADN descrito e, portanto, uma degeneração maligna acelerada. Assim, são necessários acontecimentos desfavoráveis adicionais para que o cancro se desenvolva. Este mecanismo explica a acumulação de carcinomas colorrectais em particular, uma vez que a dinâmica da formação de adenomas representa provavelmente um factor de risco independente [4].
O lobo em pele de ovelha
Mas quando é que os testes genéticos fazem sentido? Quais são os sinais clínicos que sugerem a presença da síndrome de Lynch? Em contraste com outras síndromes como a polipose adenomatosa familiar (FAP), não existem características fenotípicas claras na síndrome HNPCC que sugiram uma doença hereditária. Existem normalmente apenas adenomas ou carcinomas únicos, que são clinicamente indistinguíveis de tumores esporádicos. Só com a ocorrência de várias malignidades, devido à idade frequentemente jovem das pessoas afectadas ou a uma história familiar conspícua, se pode assim suspeitar da síndrome de Lynch [5]. Por exemplo, a idade média no diagnóstico do cancro colorrectal é de 45 anos e em cerca de 30% dos casos é adicionado outro tumor típico dentro de dez anos [6]. Em geral, os tumores associados ao HNPCC são na sua maioria adenocarcinomas; no intestino, ocorrem preferencialmente no hemicólon direito.
Para facilitar a identificação dos doentes em risco, foram desenvolvidos vários resultados de diagnóstico ao longo dos anos, nomeadamente os Critérios de Amesterdão e as Directrizes de Bethesda. Além disso, o modelo PREMM existe para o cálculo de probabilidades. O elemento central de todos estes sistemas é uma história familiar detalhada. Para preencher os critérios de Amesterdão II, que são suficientes para o diagnóstico do HNPCC, pelo menos três familiares devem ter cancro associado à síndrome de Lynch, pelo menos duas gerações consecutivas devem ser afectadas e pelo menos um membro da família deve ter tido a doença antes dos 50 anos de idade (visão geral 1) [7]. Em casos menos claros, as Directrizes de Bethesda ajudam a identificar indivíduos potencialmente afectados e a orientá-los para diagnósticos apropriados (Resumo 2). Com a crescente disponibilidade da análise de MSI, que hoje em dia já é rotineiramente realizada para muitos carcinomas, estas directrizes estão a perder o seu significado, mas ainda são importantes na avaliação clínica dos resultados [7]. Isto porque a instabilidade dos microsatélites não prova a síndrome de Lynch, pois ocorre em 10-15% de todos os cólon e 15-20% de todos os carcinomas endometriais [5].

Instabilidade por microssatélite detectada… e agora?
Se for detectada uma instabilidade por microsatélite ou uma deficiência das proteínas MMR no tumor de um paciente suspeito de ter síndrome de Lynch, há uma indicação de testes genéticos para desencadear mutações. Estes, bem como o aconselhamento genético, são serviços obrigatórios prestados pela companhia de seguros de saúde, desde que as condições para o esclarecimento genético sejam cumpridas [1]. Se for encontrada uma mutação para a qual tenha sido estabelecida uma ligação clara com a doença, devem também ser recomendados testes direccionados aos membros da família. Neste contexto, as alterações genéticas de (ainda) importância desconhecida são problemáticas. Neste caso, a prevenção baseia-se na história médica pessoal e na história familiar e não no resultado do exame genético. O procedimento é, portanto, o mesmo que no caso de um achado discreto; os membros da família não devem ser testados.
Na ausência de terapias específicas, o tratamento de tumores malignos baseia-se principalmente nas recomendações que também se aplicam aos tumores esporádicos. Em fases avançadas, porém, os inibidores de pontos de controlo são cada vez mais utilizados. Por exemplo, o pembrolizumab é aprovado como monoterapia para o tratamento de primeira linha do cancro colorrectal metastático com MSI elevado ou reparação de desajustes de ADN defeituosos [8]. No que diz respeito à utilização de mais imunoterapêutica e ao valor da instabilidade dos microssatélites como marcador preditivo, podem esperar-se dados interessantes nos próximos anos.
Prevenção e detecção precoce
As provas genéticas da síndrome de Lynch resultam numa longa cauda de ratazana de recomendações para um maior rastreio do cancro. Estes também se aplicam aos familiares portadores da mutação correspondente. Para além de um estilo de vida saudável e de medidas de detecção precoce, são também utilizadas cirurgias preventivas e medicação. As colonoscopias regulares e os exames ginecológicos devem prevenir o cancro colorrectal e endometrial. Recomenda-se também o rastreio de Helicobacter pylori (Visão Geral 3). Não existe actualmente uma detecção precoce eficaz do carcinoma ovariano, que também ocorre frequentemente, ou de tumores uroteliais. Isto torna ainda mais importante a sensibilização das pessoas afectadas aos sintomas correspondentes e, dependendo da doença tumoral na família, a implementação de medidas adicionais de monitorização.
Em alguns casos, uma histerectomia profiláctica com ou sem adnexectomia é realizada após o planeamento familiar ter sido concluído. Também são por vezes utilizadas ressecções prolongadas do cólon. O resultado final é que o benefício da cirurgia preventiva da síndrome de Lynch não é claro e deve ser pesado numa base individual [1]. Há também falta de dados fiáveis sobre a utilização profiláctica da aspirina, especialmente sobre a dosagem e a duração da administração. De acordo com os resultados a longo prazo do estudo CAPP II, a ingestão diária de 600 mg de ácido acetilsalicílico reduz significativamente o risco de todas as malignidades associadas ao HNPCC após um período de latência de cerca de quatro anos. Este efeito parece durar cerca de dez anos após dois anos de administração, mesmo sem mais administração [9]. Com o CAPP III, está actualmente em curso um estudo para avaliar doses mais baixas.
Embora muito tenha sido feito no diagnóstico genético e na gestão de pacientes com síndrome de Lynch nos últimos anos, especialmente nos centros, é provável que a síndrome ainda esteja subdiagnosticada [10]. A fim de contribuir significativamente para um melhor tratamento das pessoas afectadas, podemos fazer uma coisa acima de tudo: Pense sobre isso.
Literatura:
- SAKK: Guia de Aconselhamento Síndrome de Lynch 2021. www.sakk.ch/sites/default/files/2020-11/Leitfaden%20Lynch-Syndrom.pdf. (último acesso 08.05.2021)
- UKB, Institute of Human Genetics: HNPCC / Síndrome de Lynch. (último acesso 08.05.2021)
- Prospective Lynch Syndrome Database (PLSD) – risco cumulativo de cancro por idade, variante genética, e sexo em portadores sujeitos a colonoscopia. www.plsd.eu.
- Engel C, et al: Eficácia da vigilância colonoscópica anual em indivíduos com cancro colorrectal hereditário não-polipose. Clin Gastroenterol Hepatol. 2010; 8(2): 174-182.
- Steinke V, et al: Cancro colorrectal hereditário sem polipose. Dtsch Arztebl Internacional. 2013; 110(3): 32-8.
- Lynch HT, et al: Revisão da síndrome de Lynch: história, genética molecular, rastreio, diagnóstico diferencial, e ramificações médico-legais. Clin Genet. 2009; 76(1): 1-18.
- Livstone EM: Síndrome de Lynch 2019. Manual MSD. www.msdmanuals.com/de/profi/gastrointestinale-erkrankungen/tumoren-des-gastrointestinaltrakts/lynch-syndrom (último acesso 08.05.2021)
- Informação sobre medicamentos da Swissmedic, a Agência Suíça para Produtos Terapêuticos. www.swissmedicinfo.ch (último acesso 08.05.2021)
- Burn J, et al: Prevenção do cancro com aspirina em cancro colorrectal hereditário (síndrome de Lynch), seguimento de 10 anos e dados de 20 anos baseados em registos no estudo CAPP2: um ensaio duplo-cego, aleatorizado e controlado por placebo. A Lanceta. 2020; 395(10240): 1855-1863.
- Morrow A, et al: Understanding implementation success: protocol for an in-depth, mixed-methods process evaluation of a cluster randomised controlled trial testing methods to improve detection of Lynch syndrome in Australian hospitals. BMJ Aberto. 2020; 10(6): e033552.
- Vasen HF, et al: Novos critérios clínicos para o cancro colorrectal hereditário não-polipose (HNPCC, síndrome de Lynch) propostos pelo grupo International Collaborative sobre HNPCC. Gastroenterologia. 1999; 116(6): 1453-1456.
- Umar A, et al: Directrizes revistas de Bethesda para o cancro colorrectal hereditário não-polipose (síndrome de Lynch) e instabilidade por micro-satélite. J Natl Cancer Inst. 2004; 96(4): 261-268.
InFo ONCOLOGy & HEMATOLOGy 2021; 9(3): 37-39